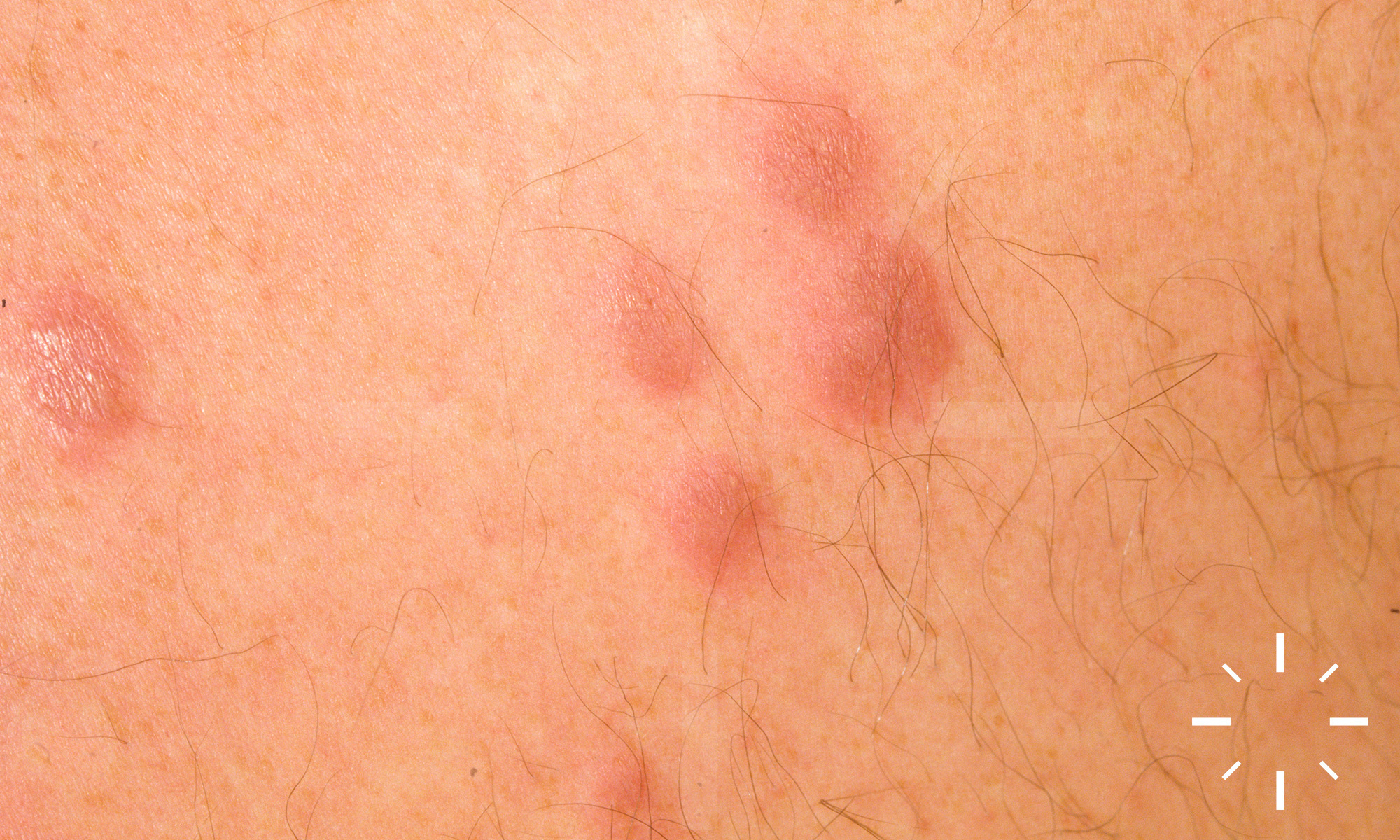
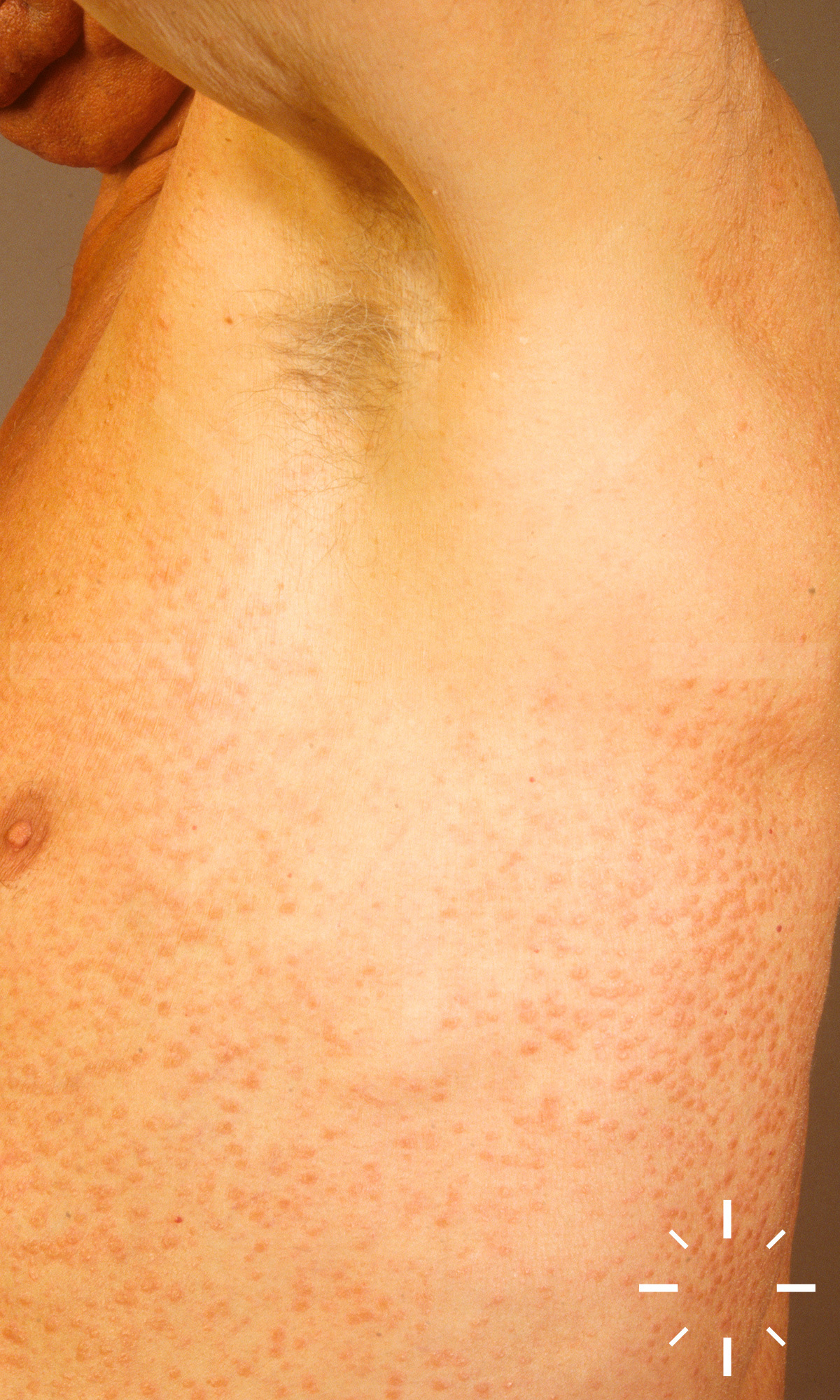
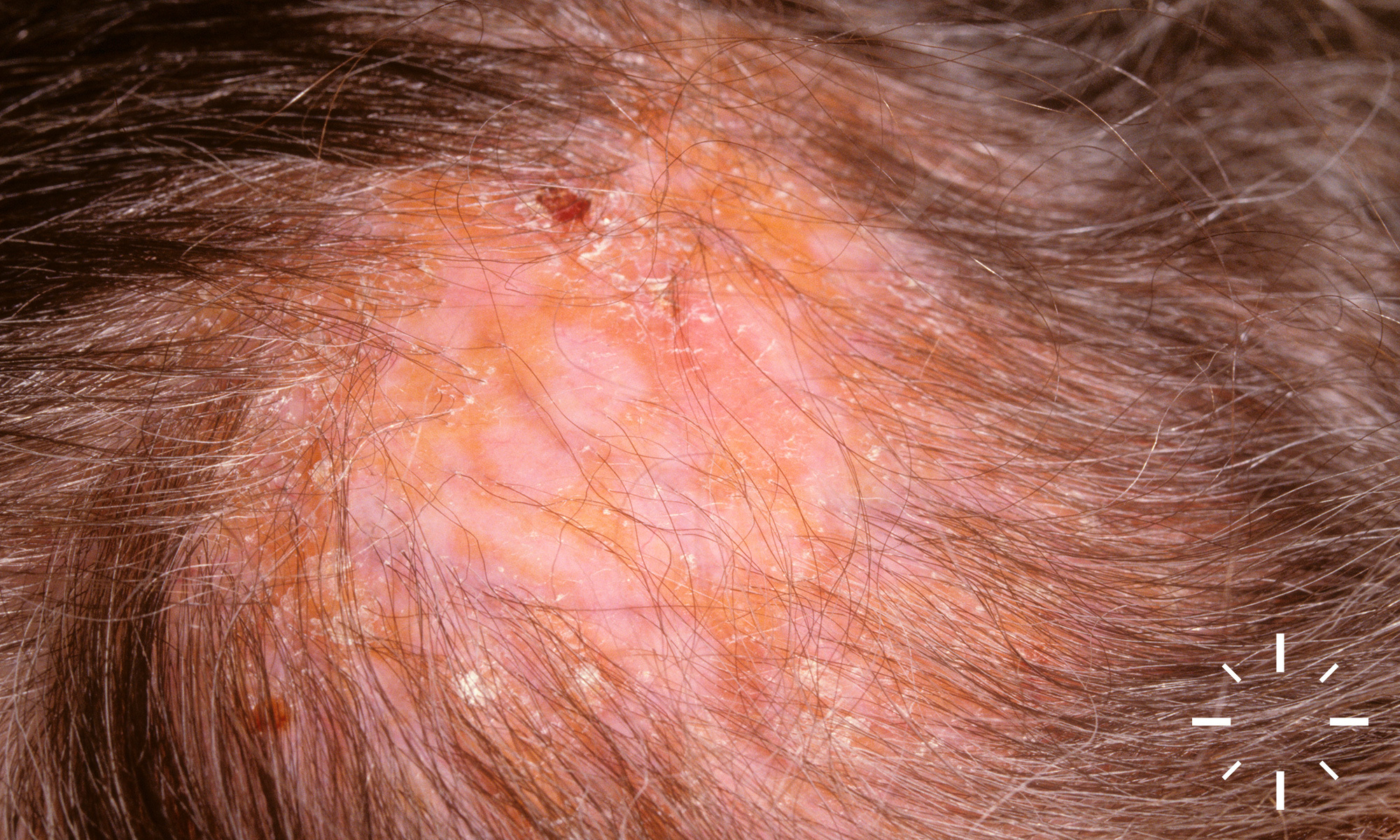
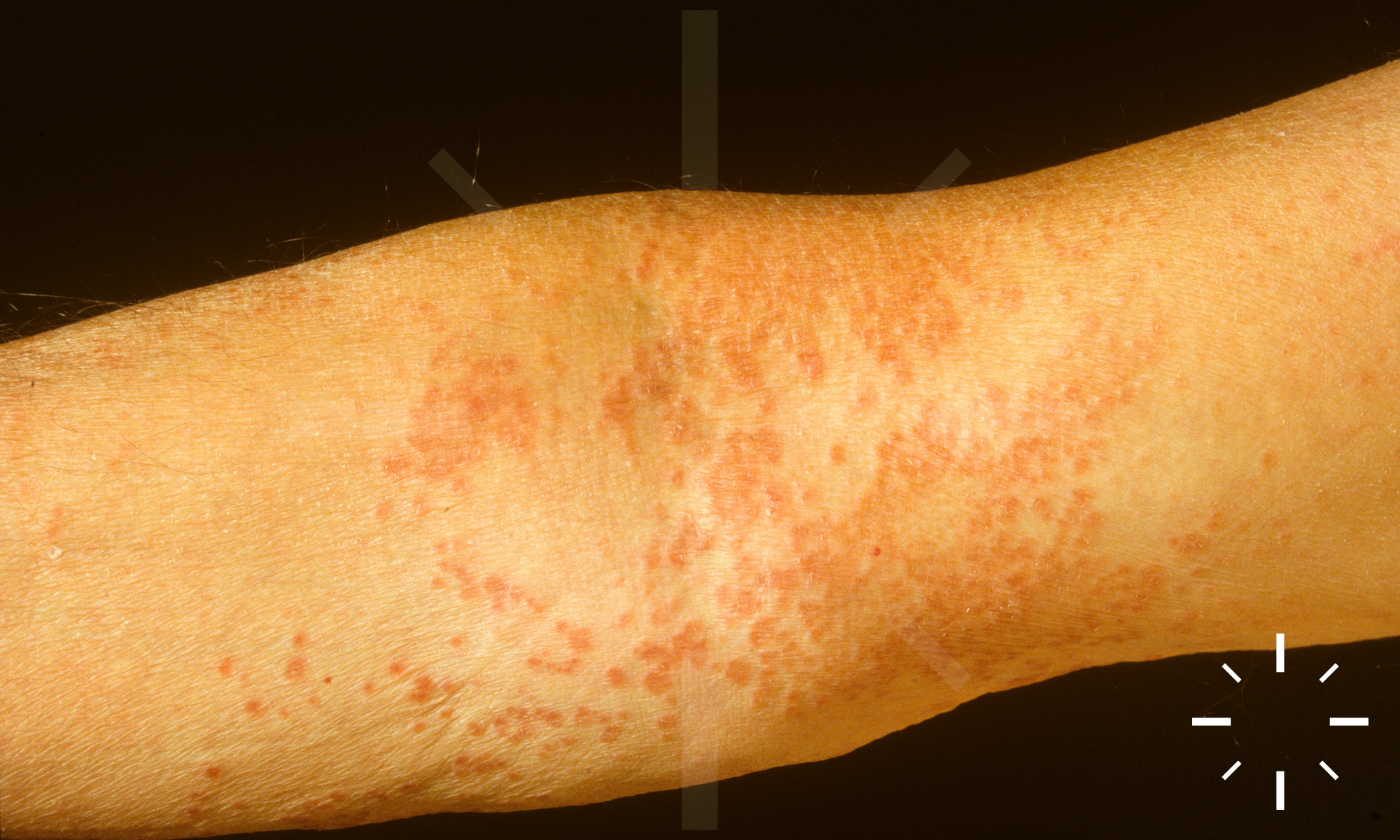
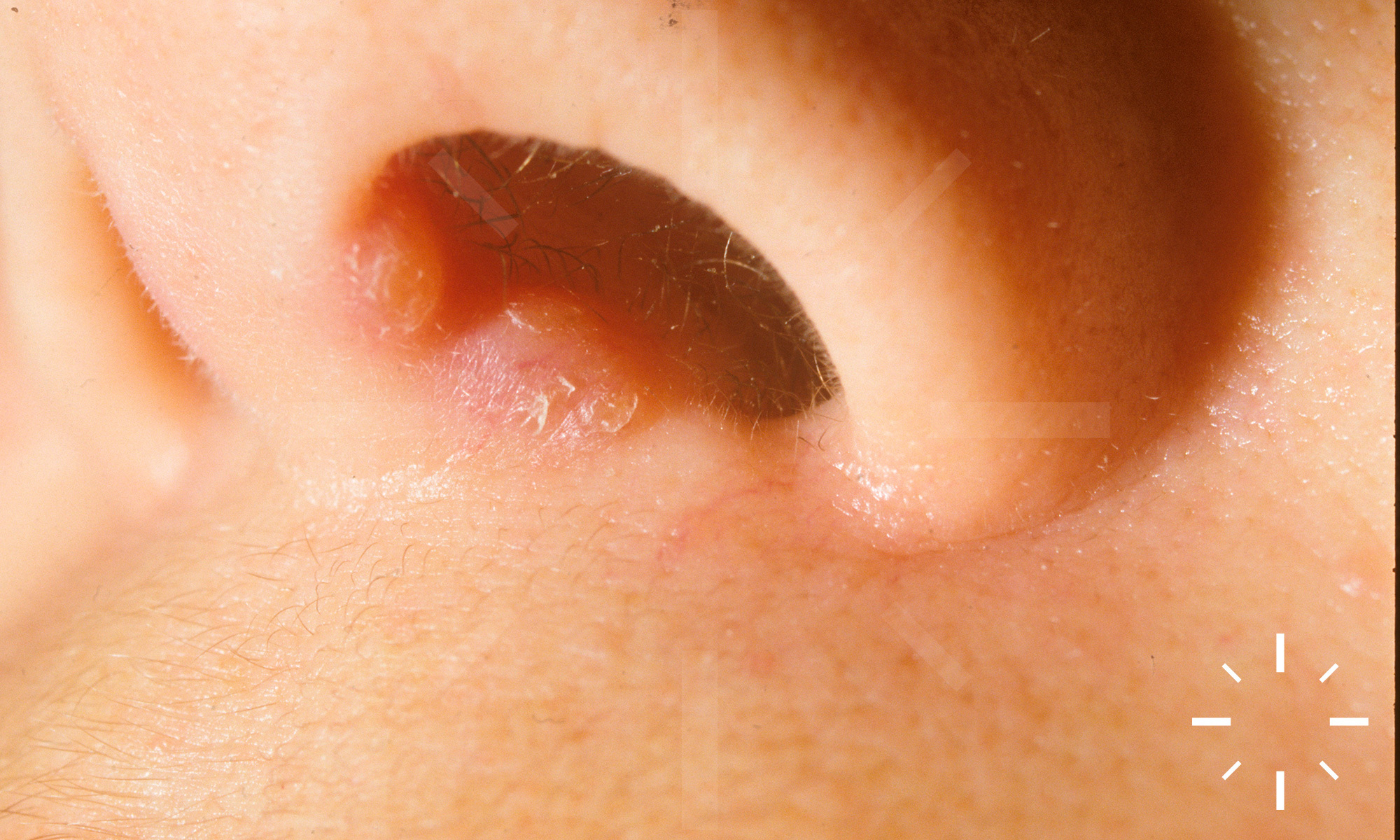
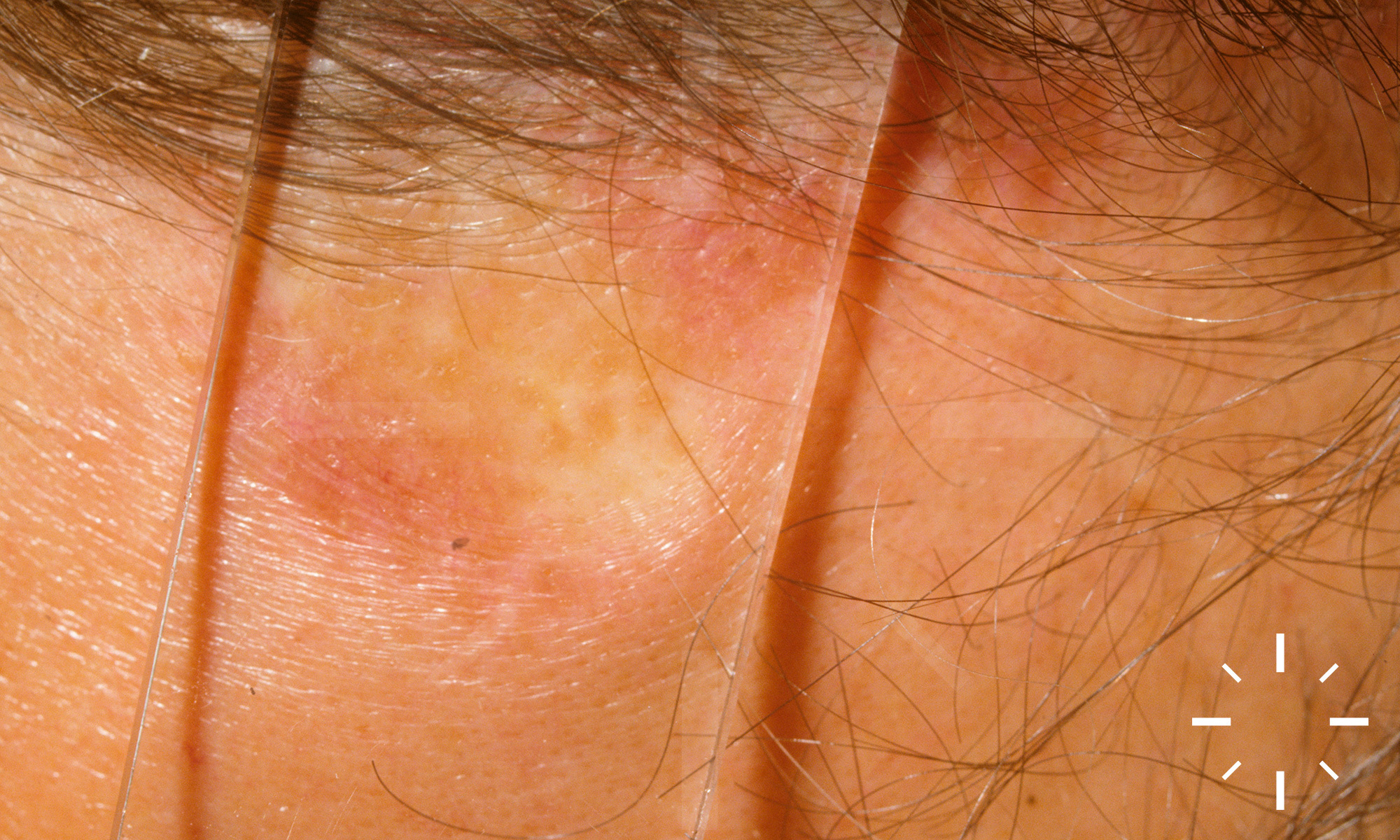
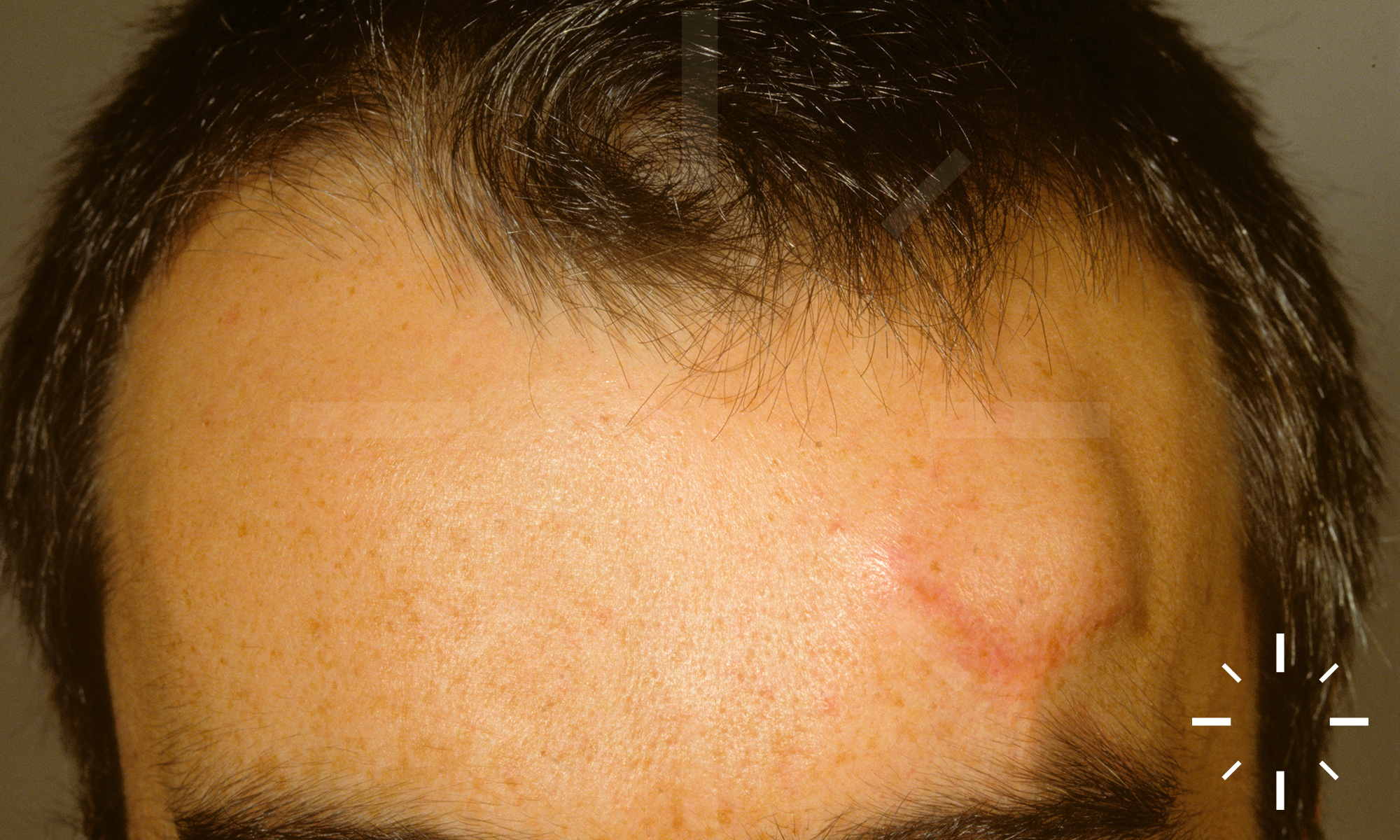
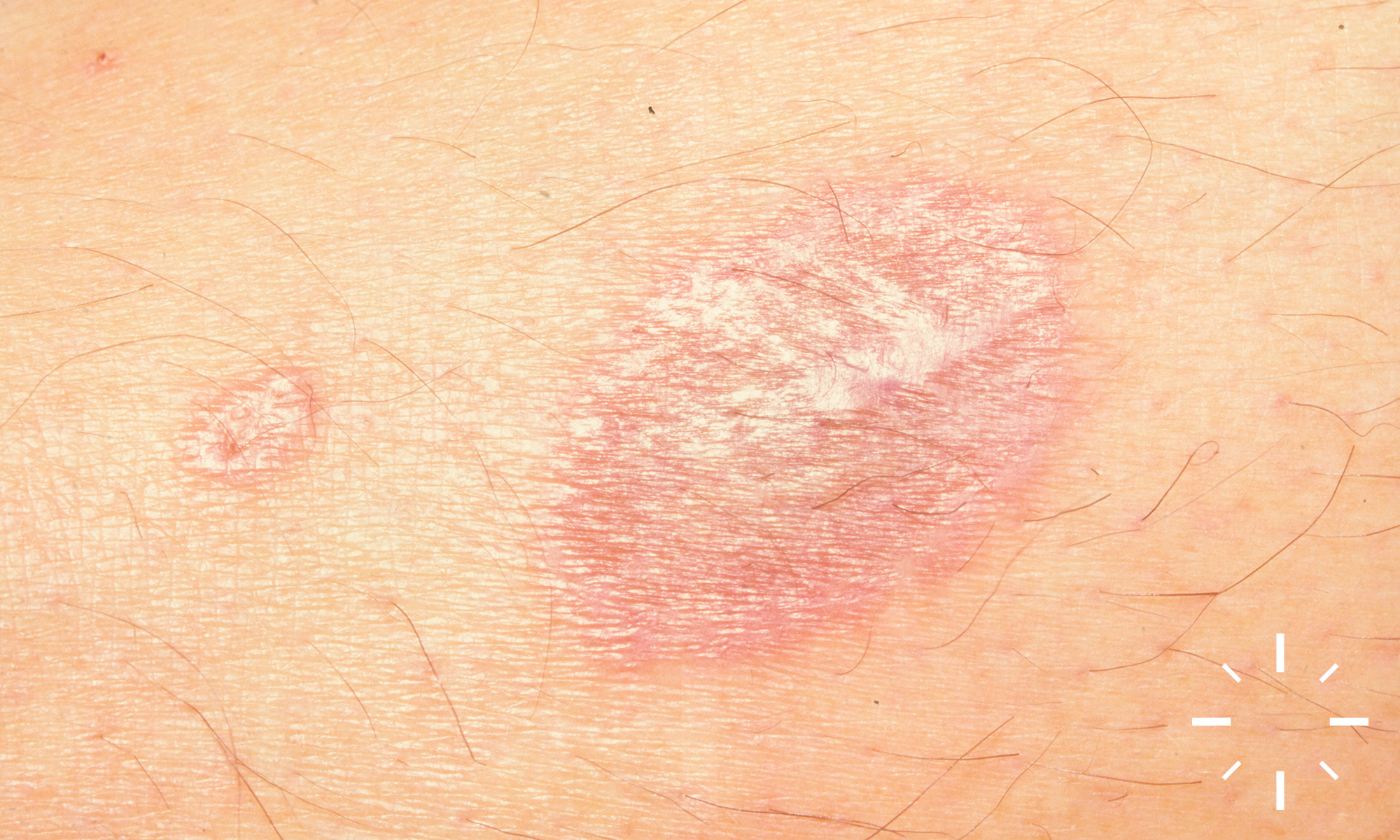
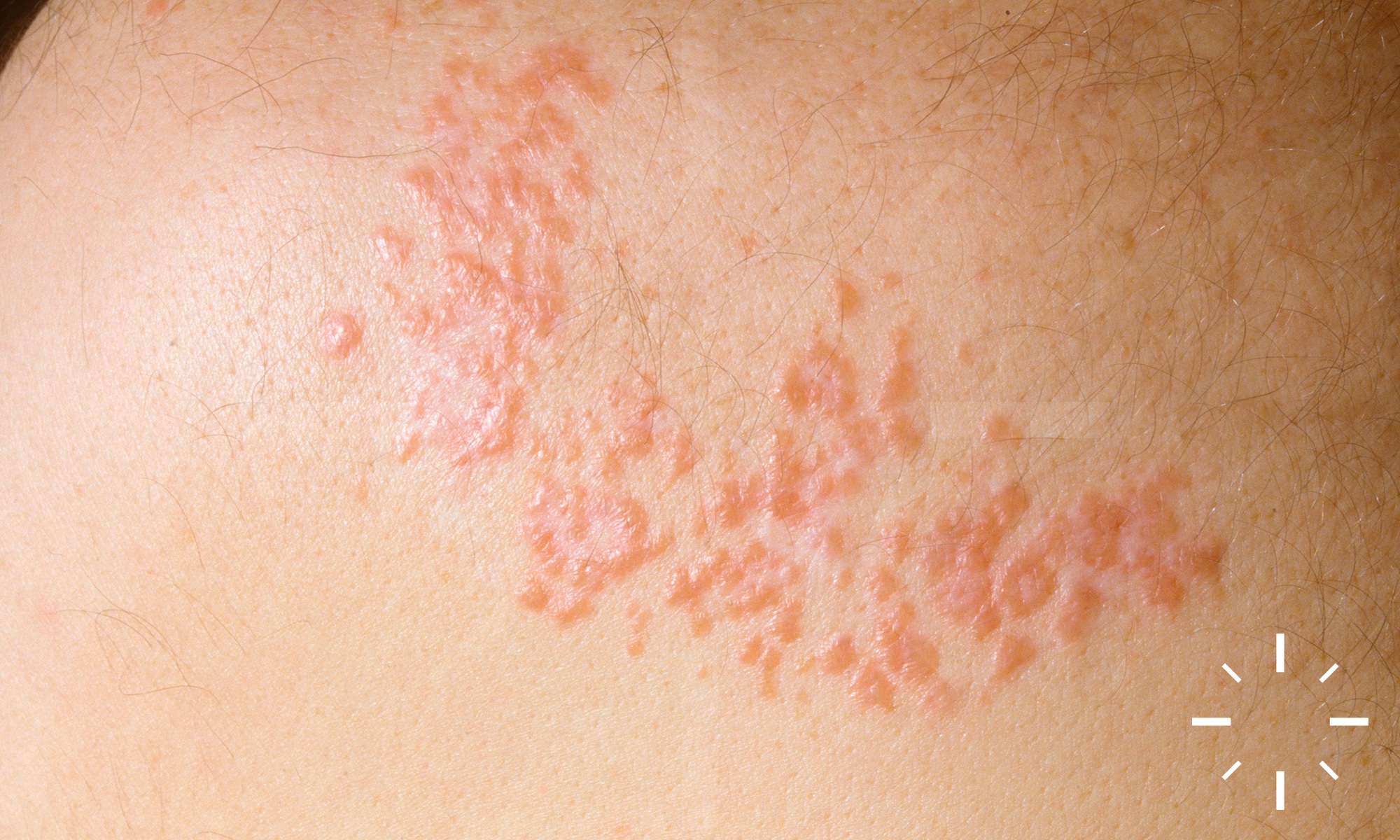
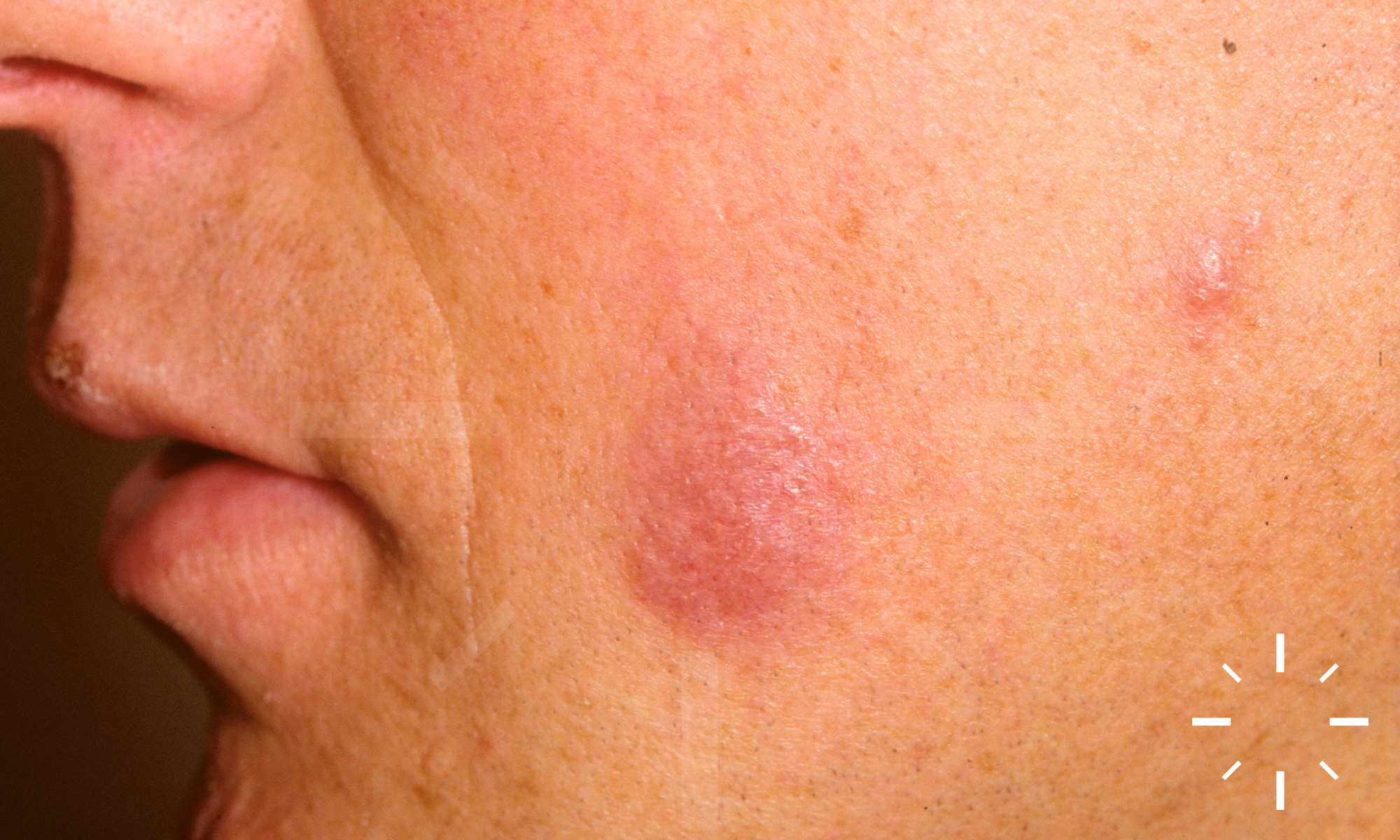
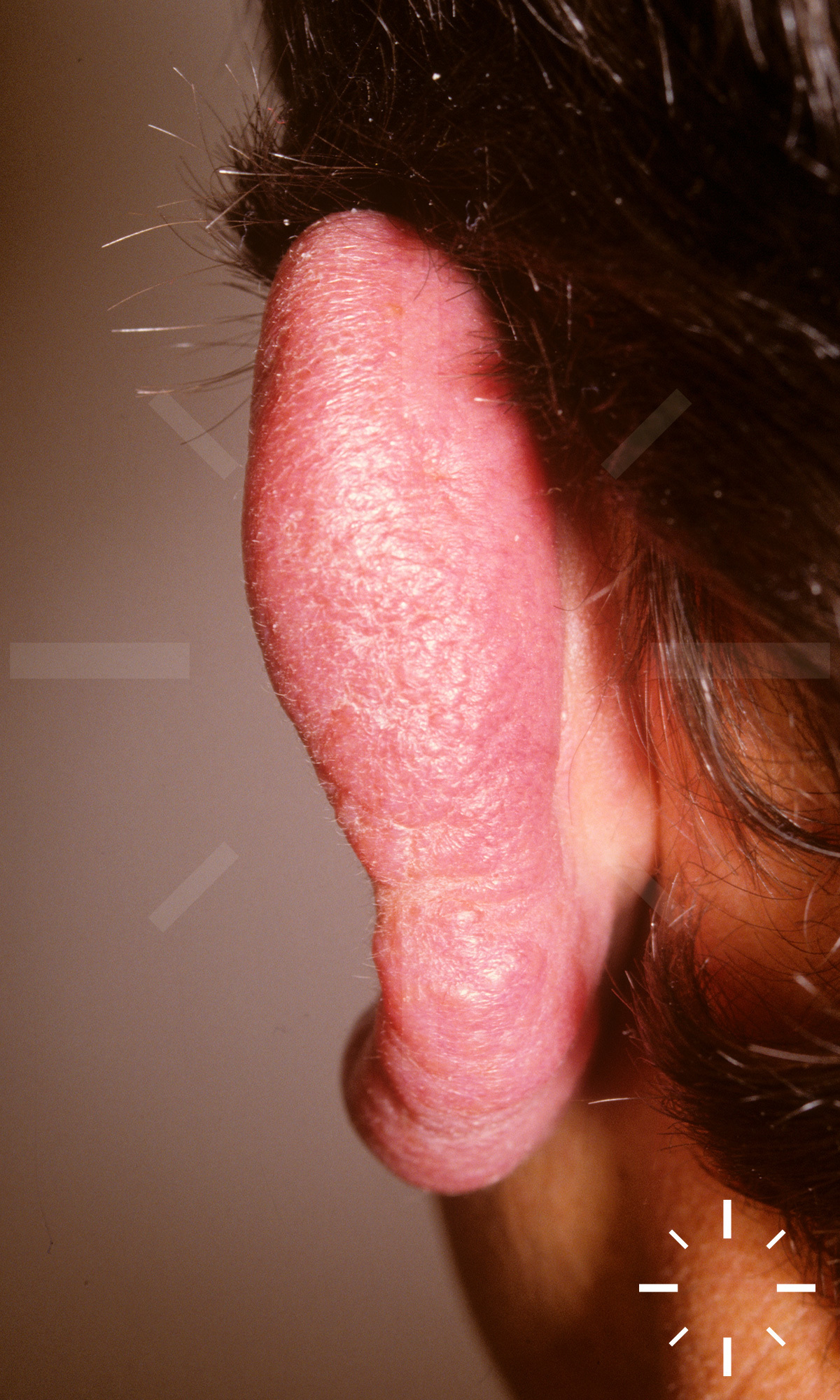
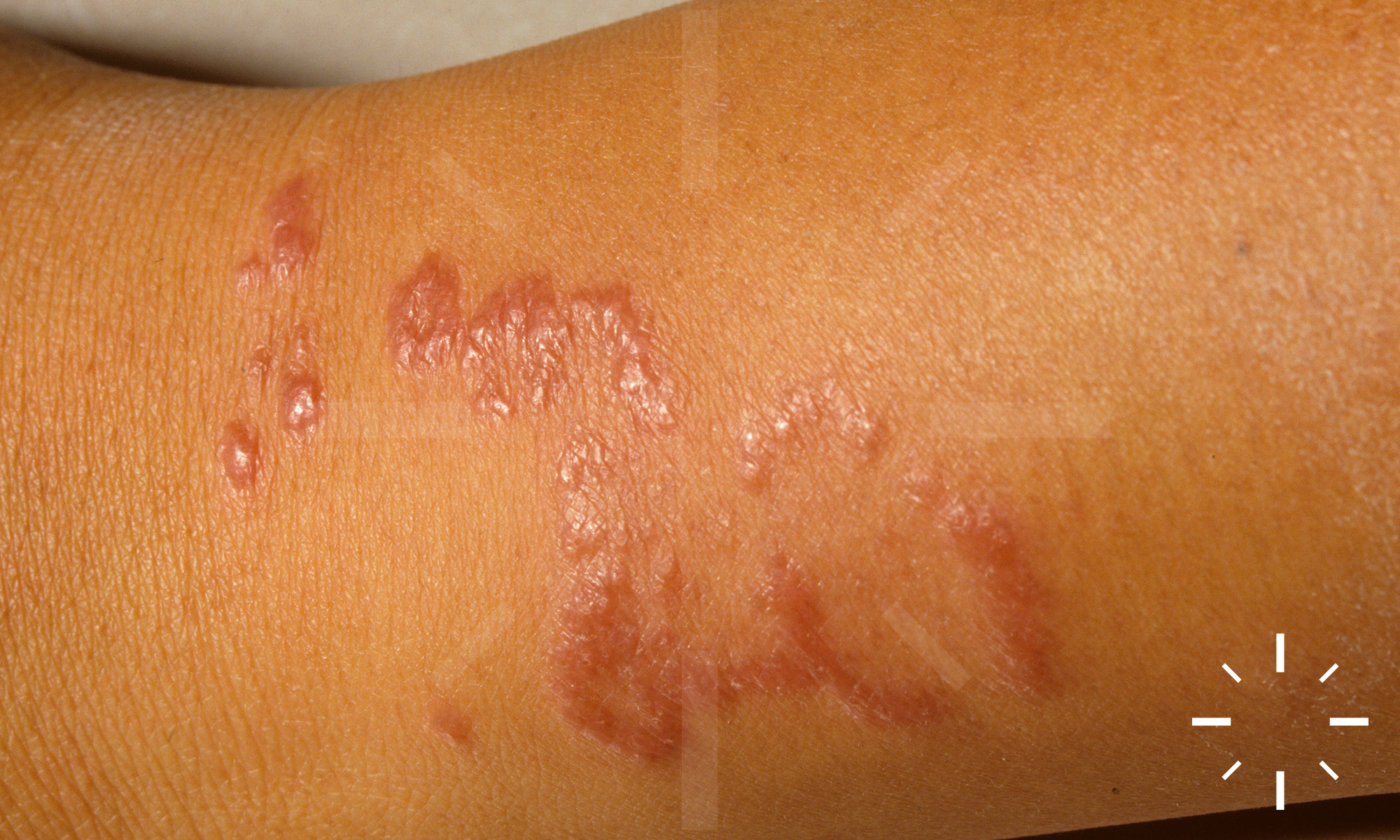
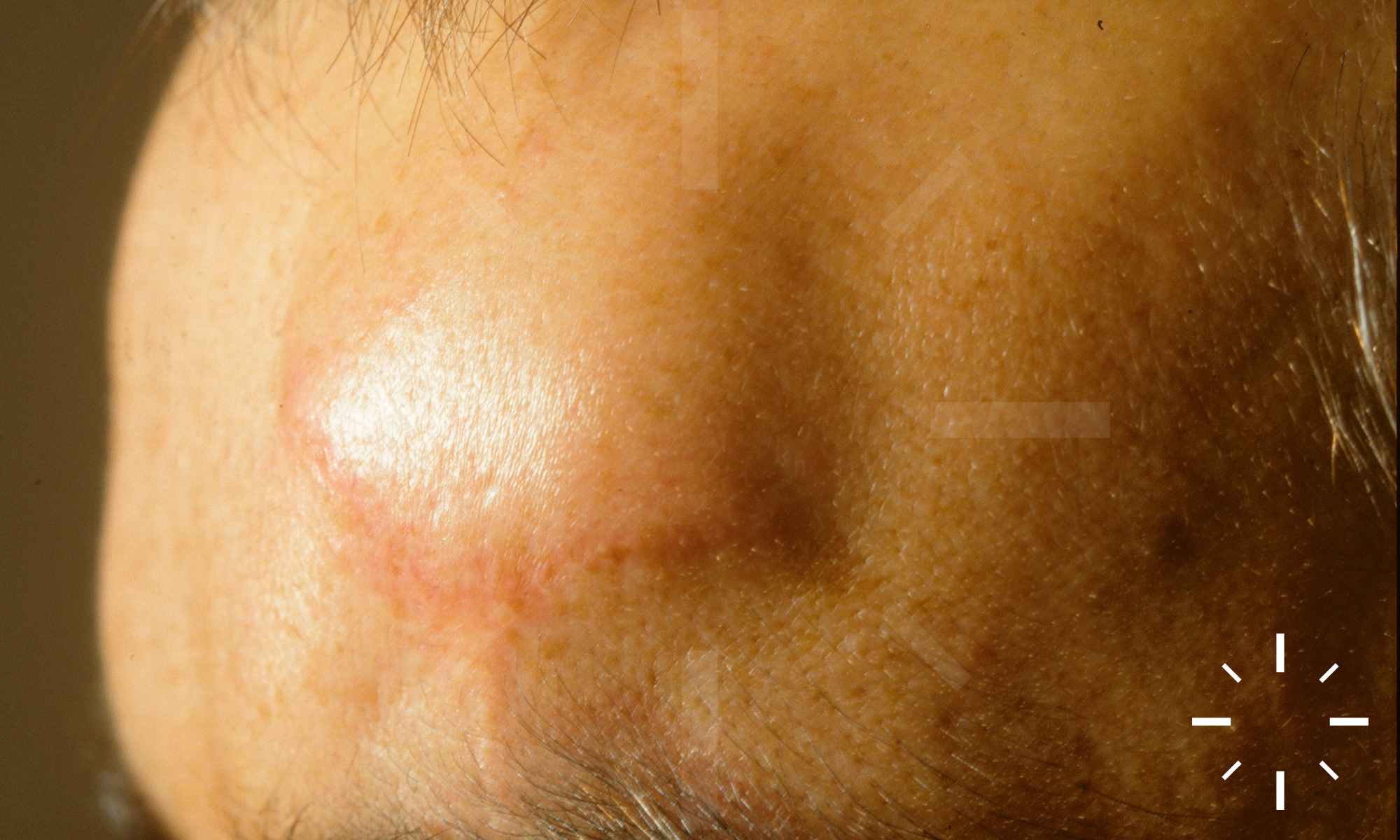
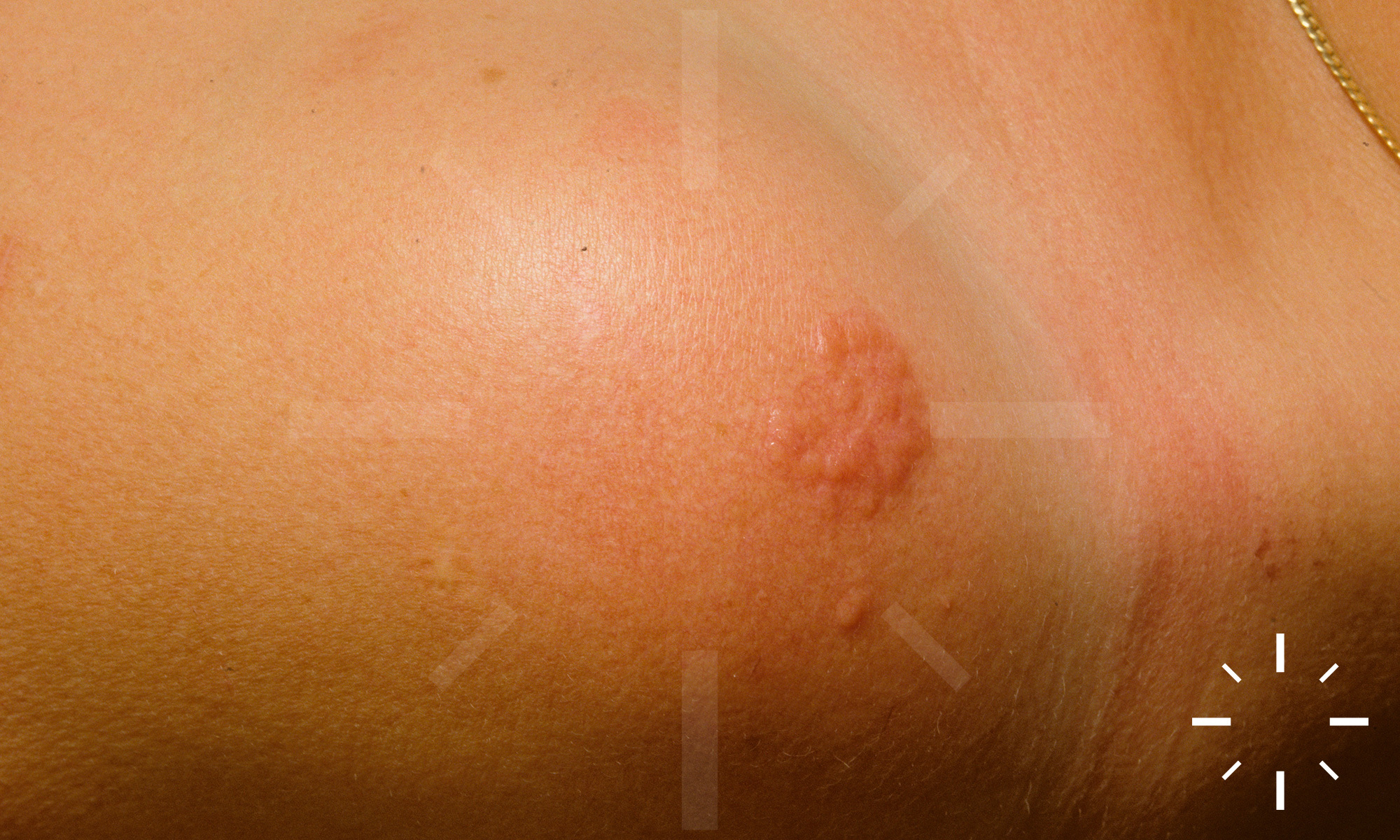
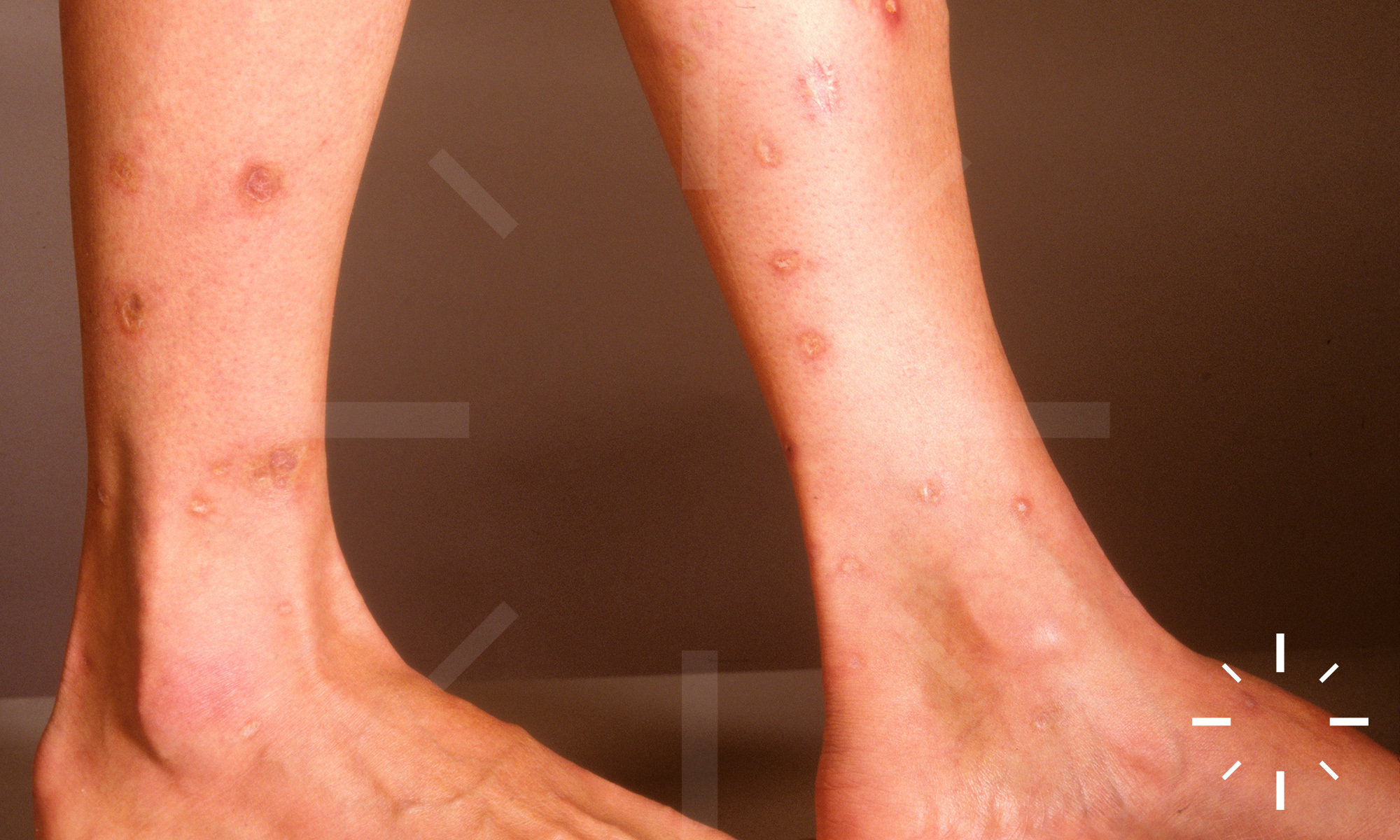
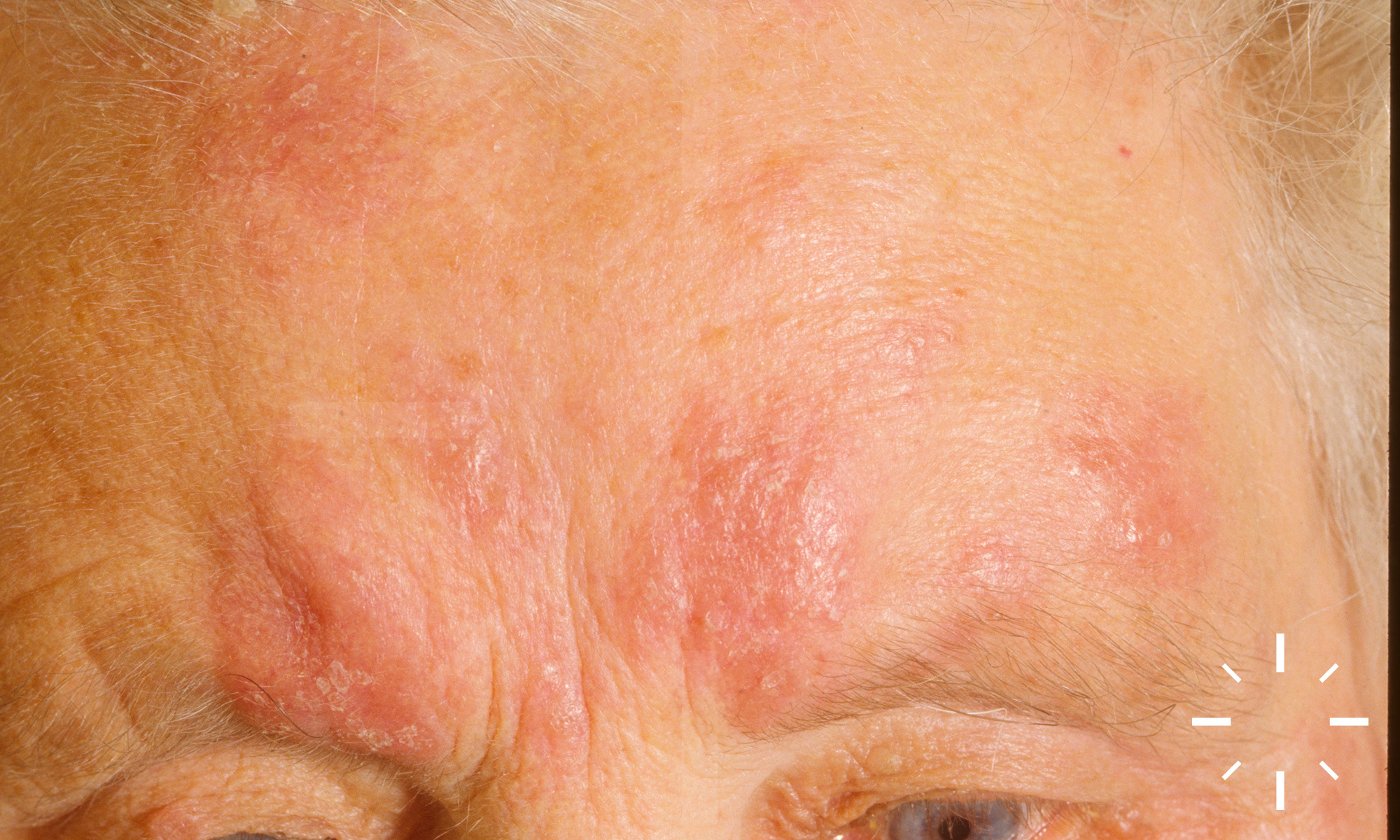
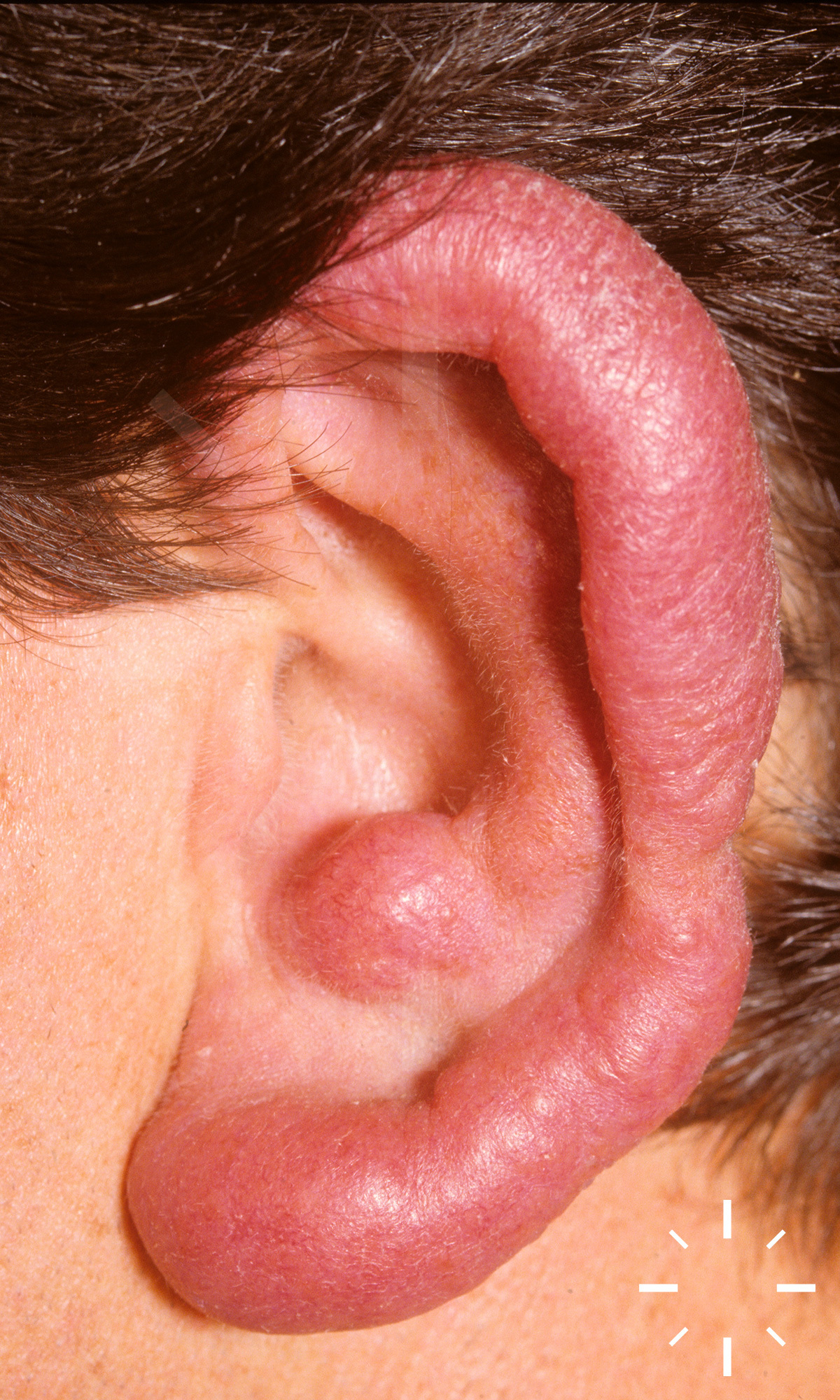
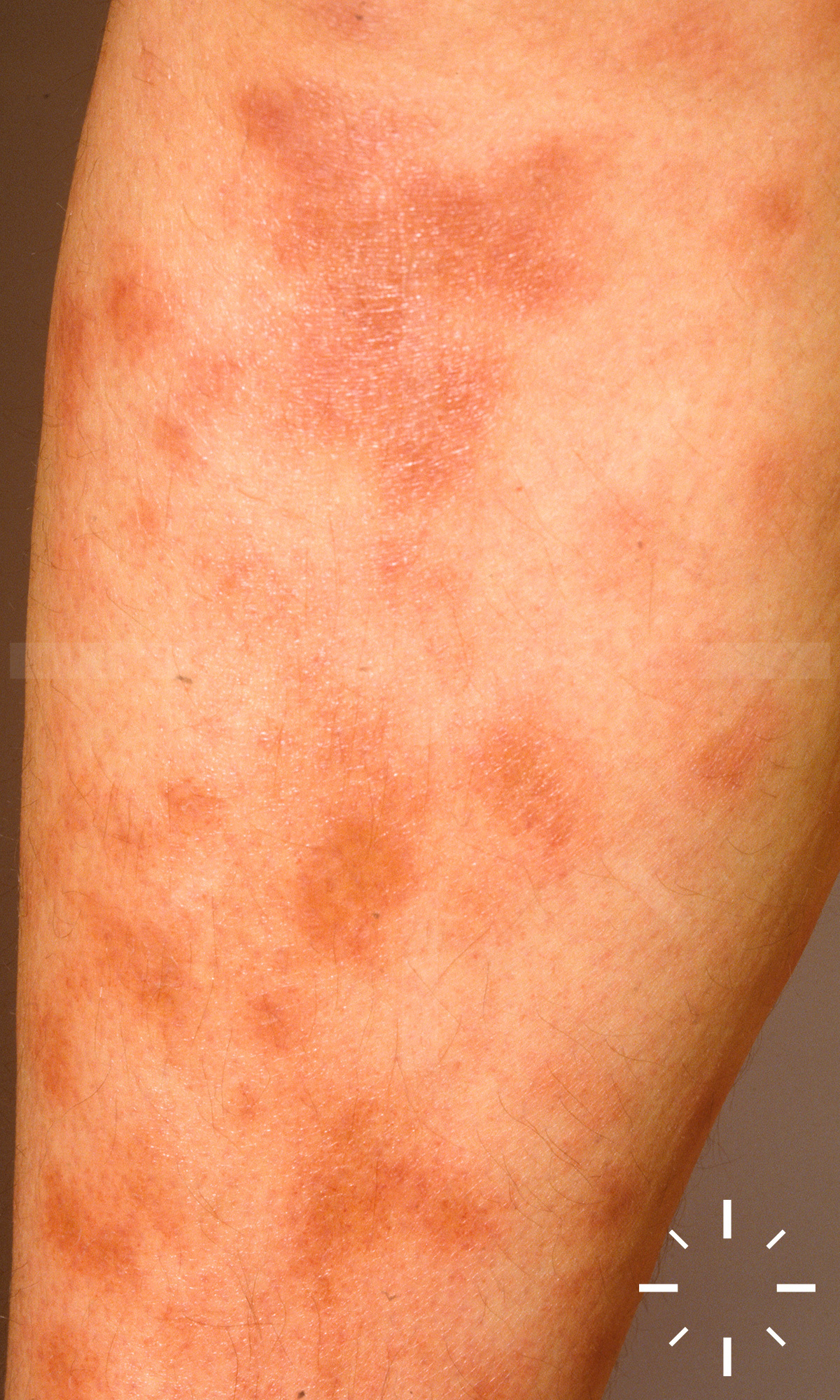
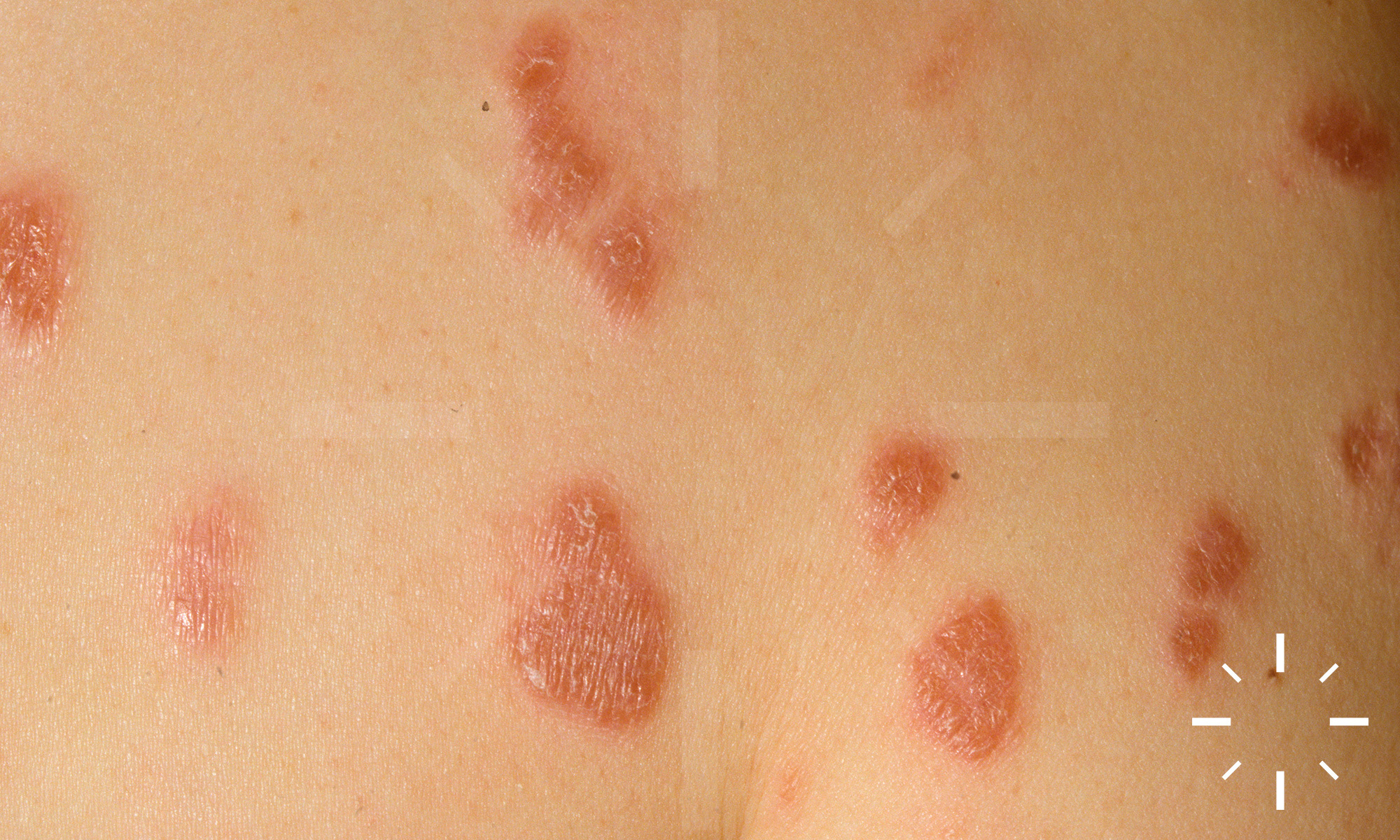
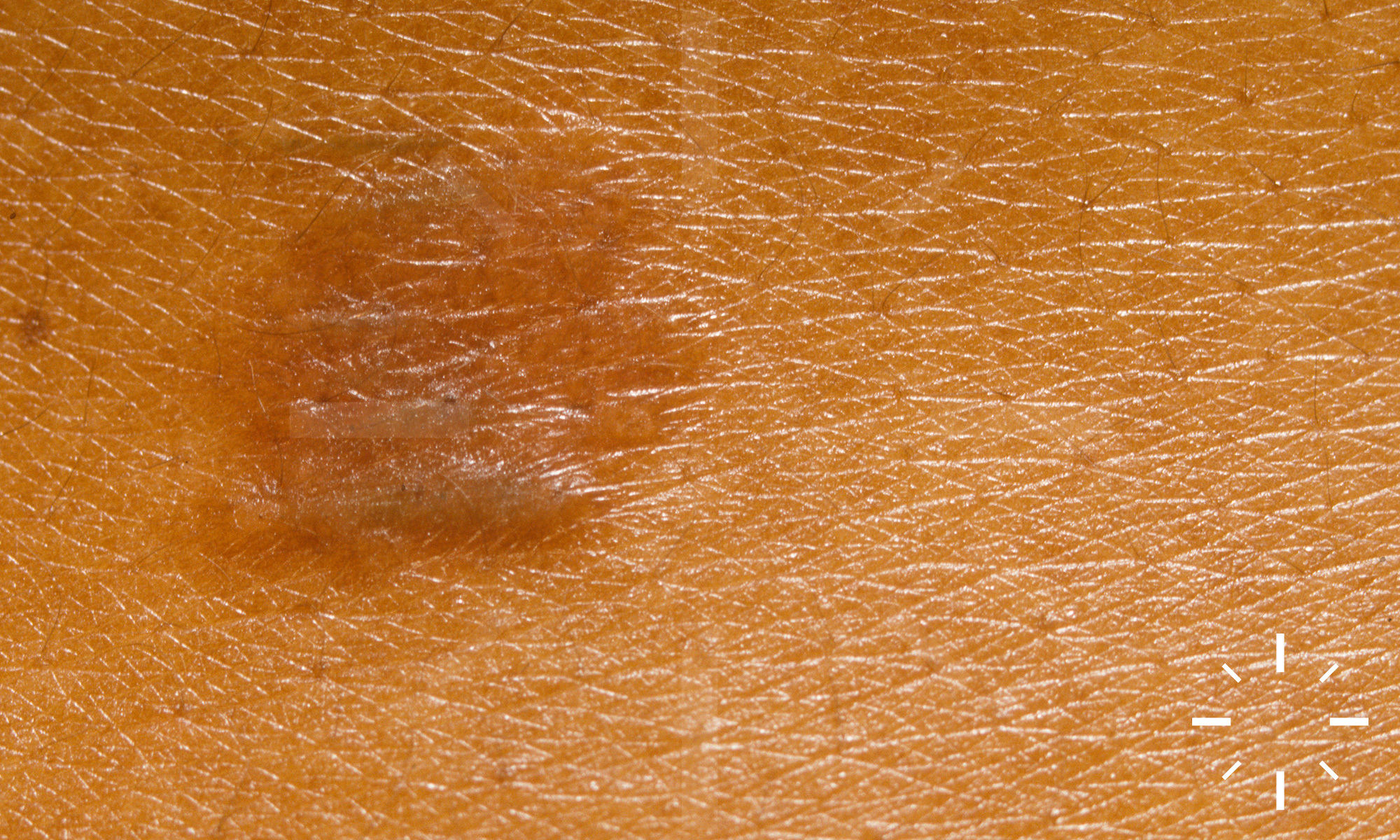
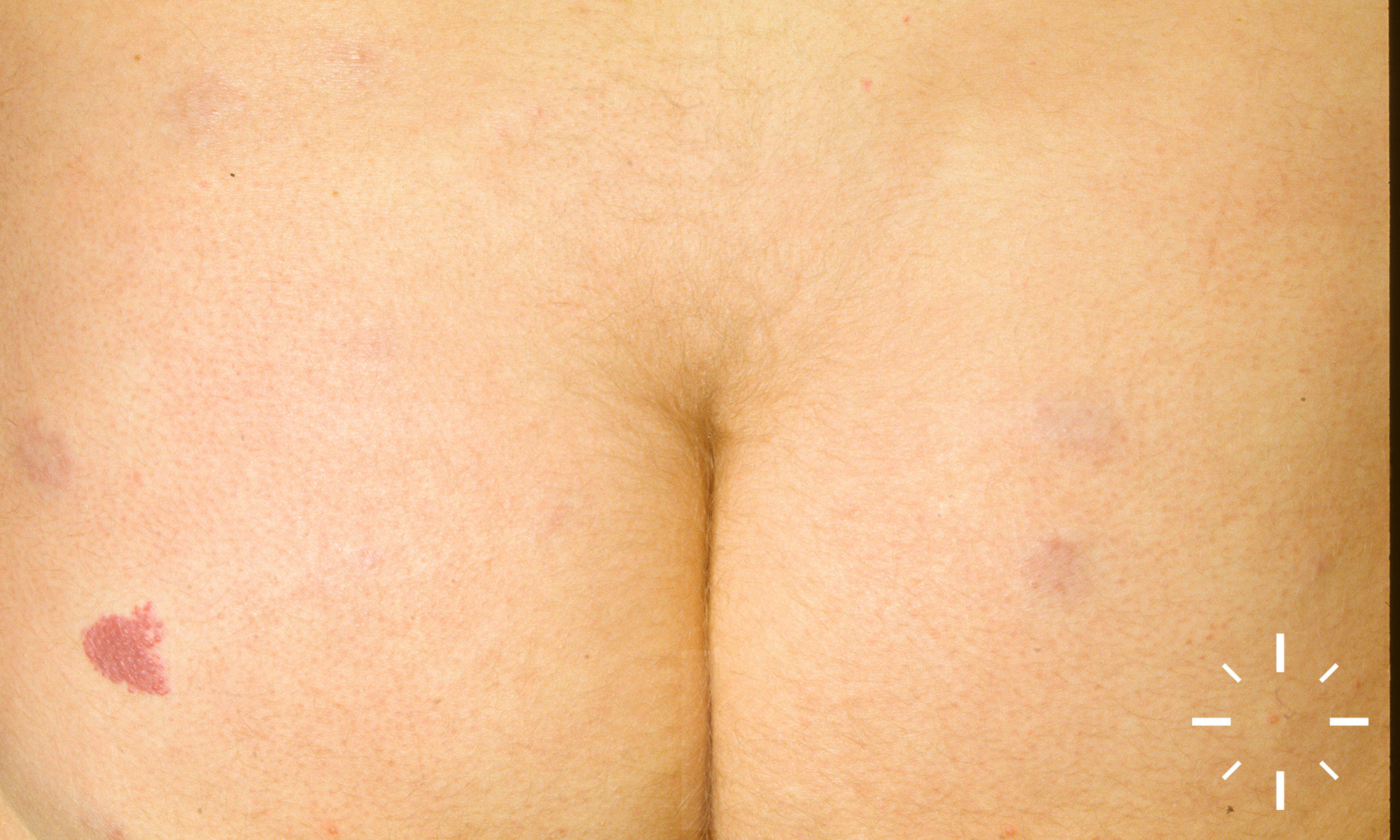
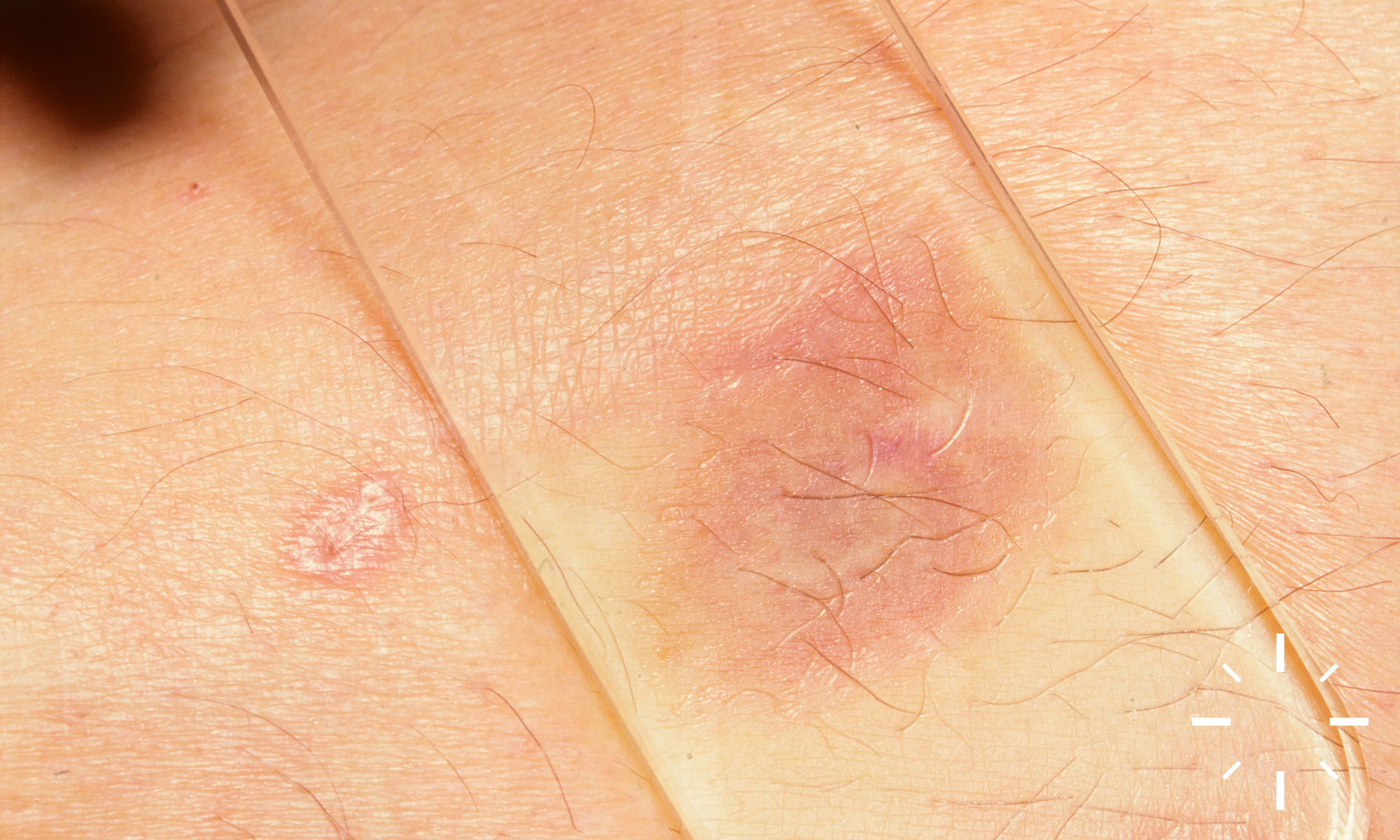
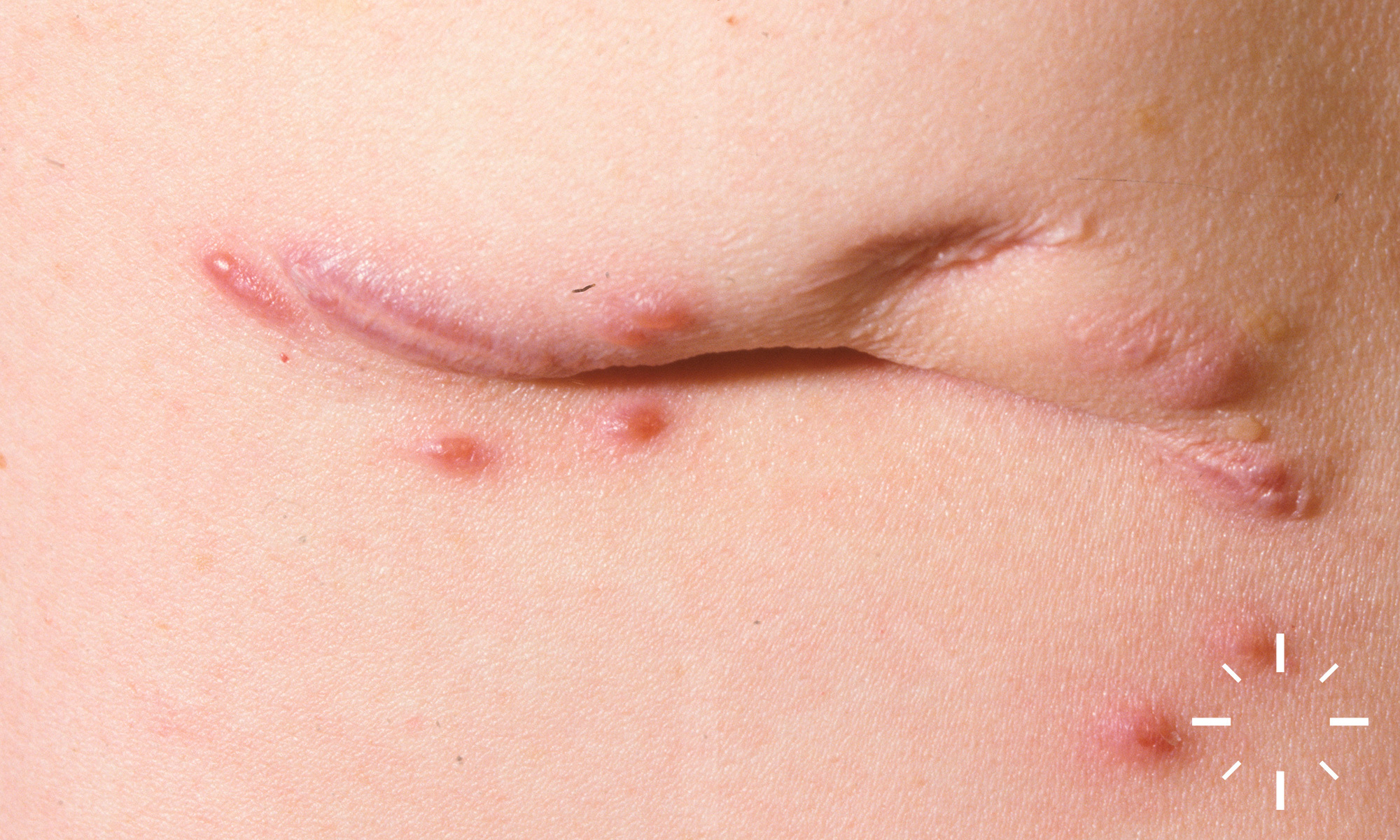
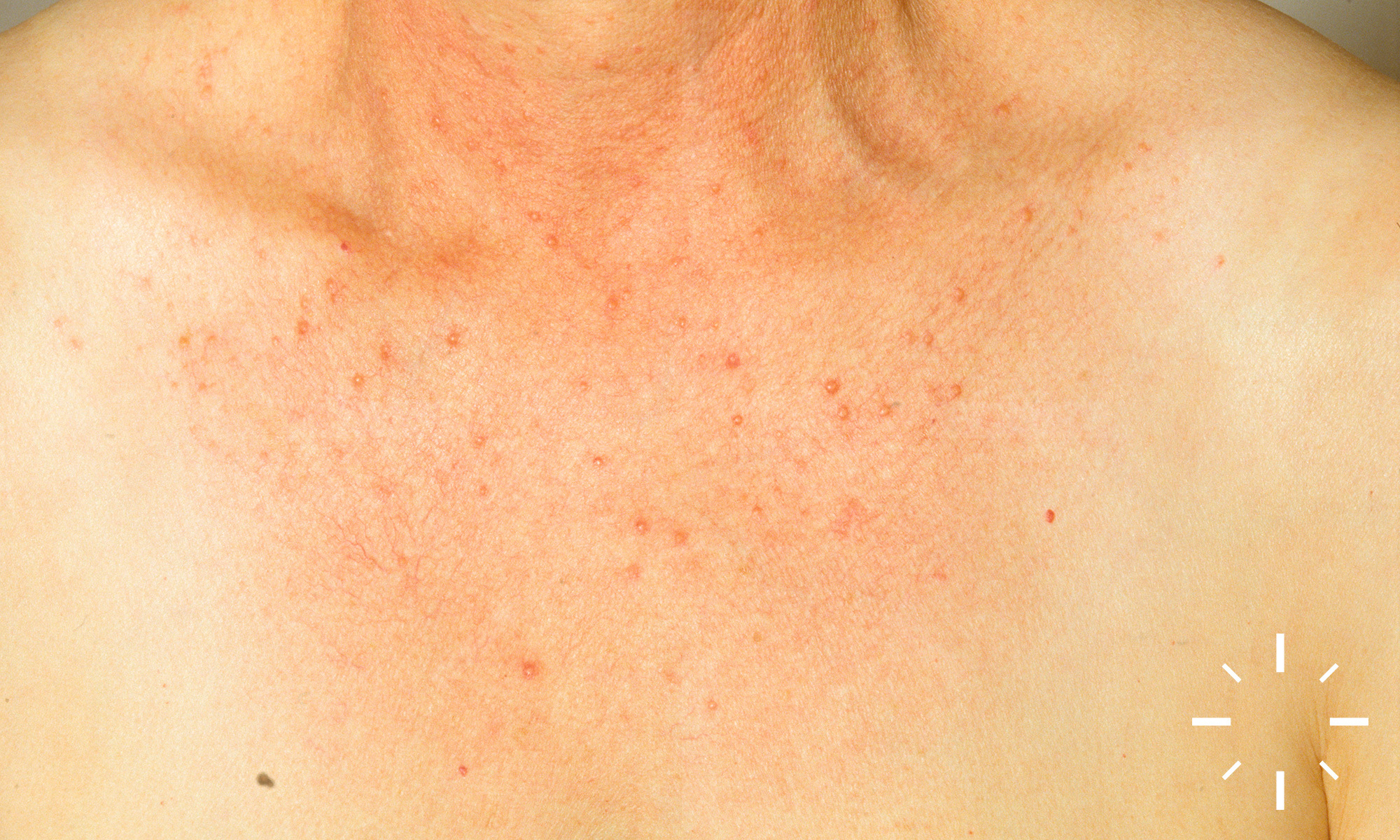
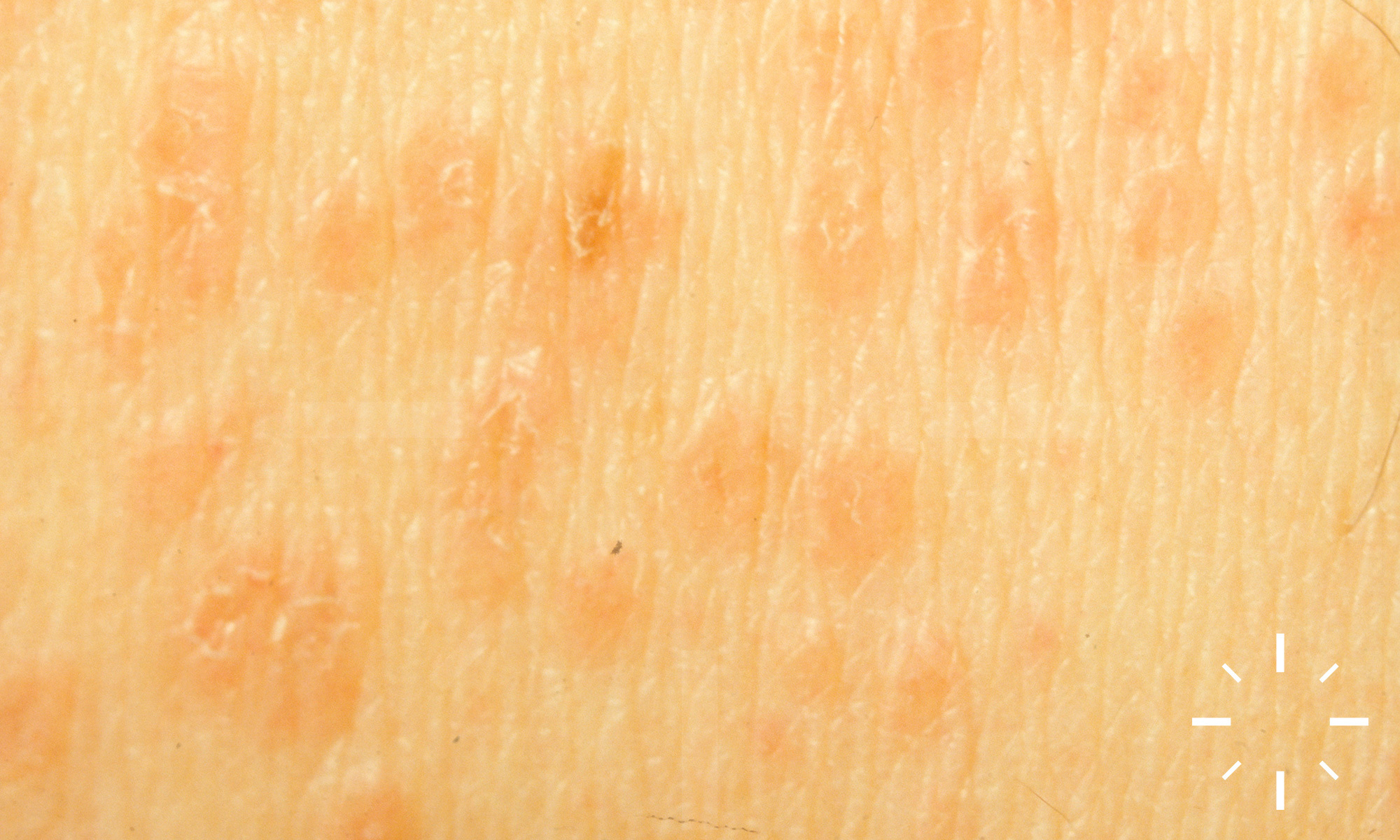
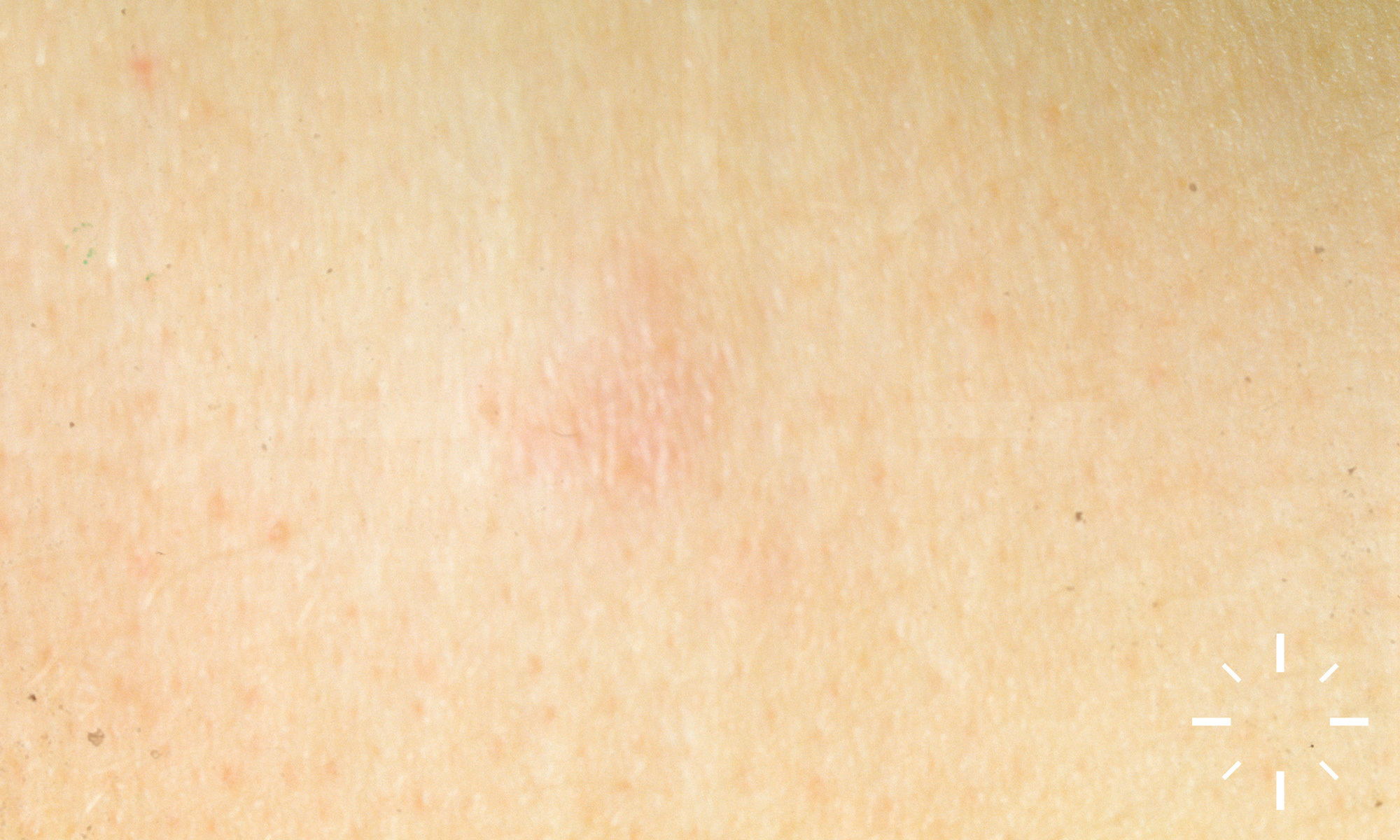
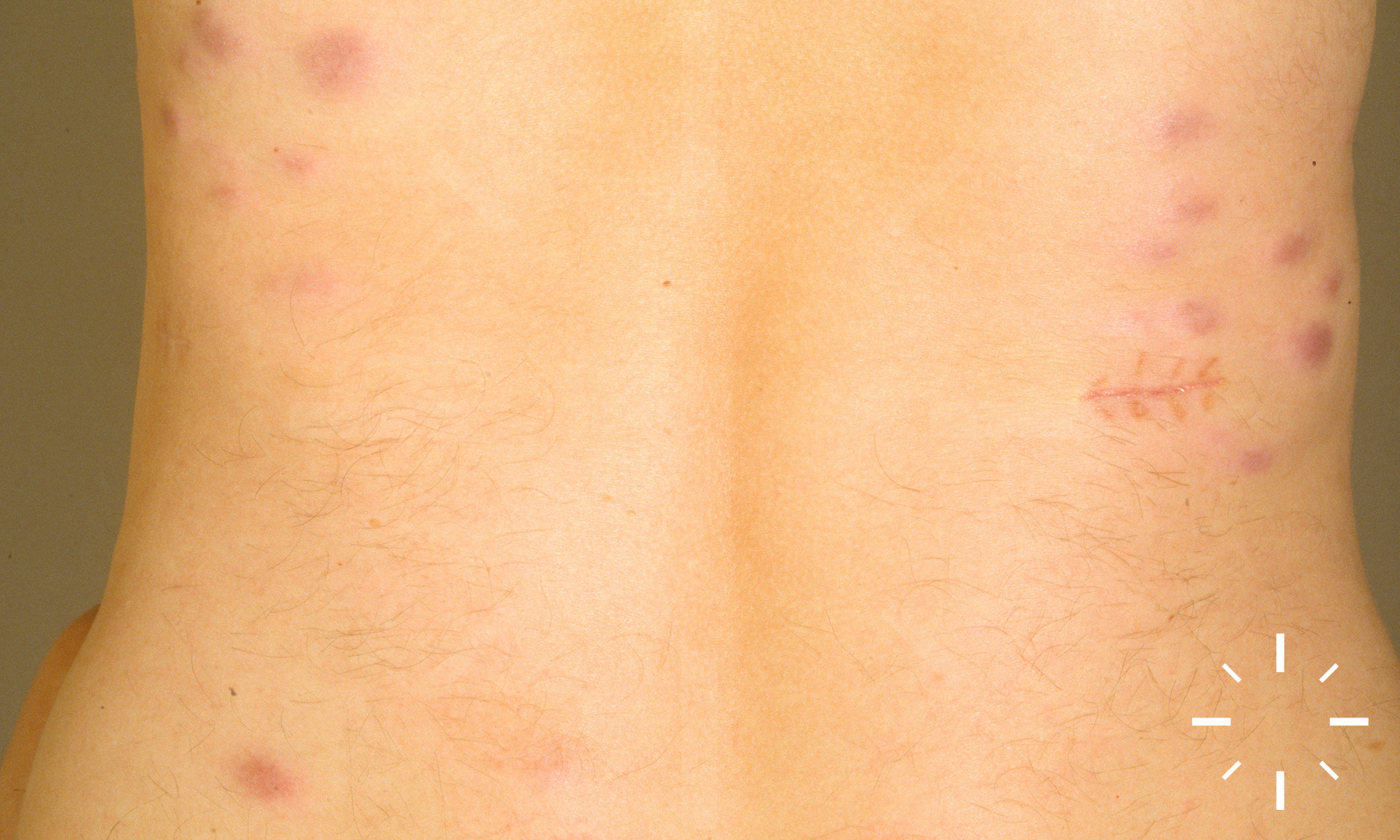
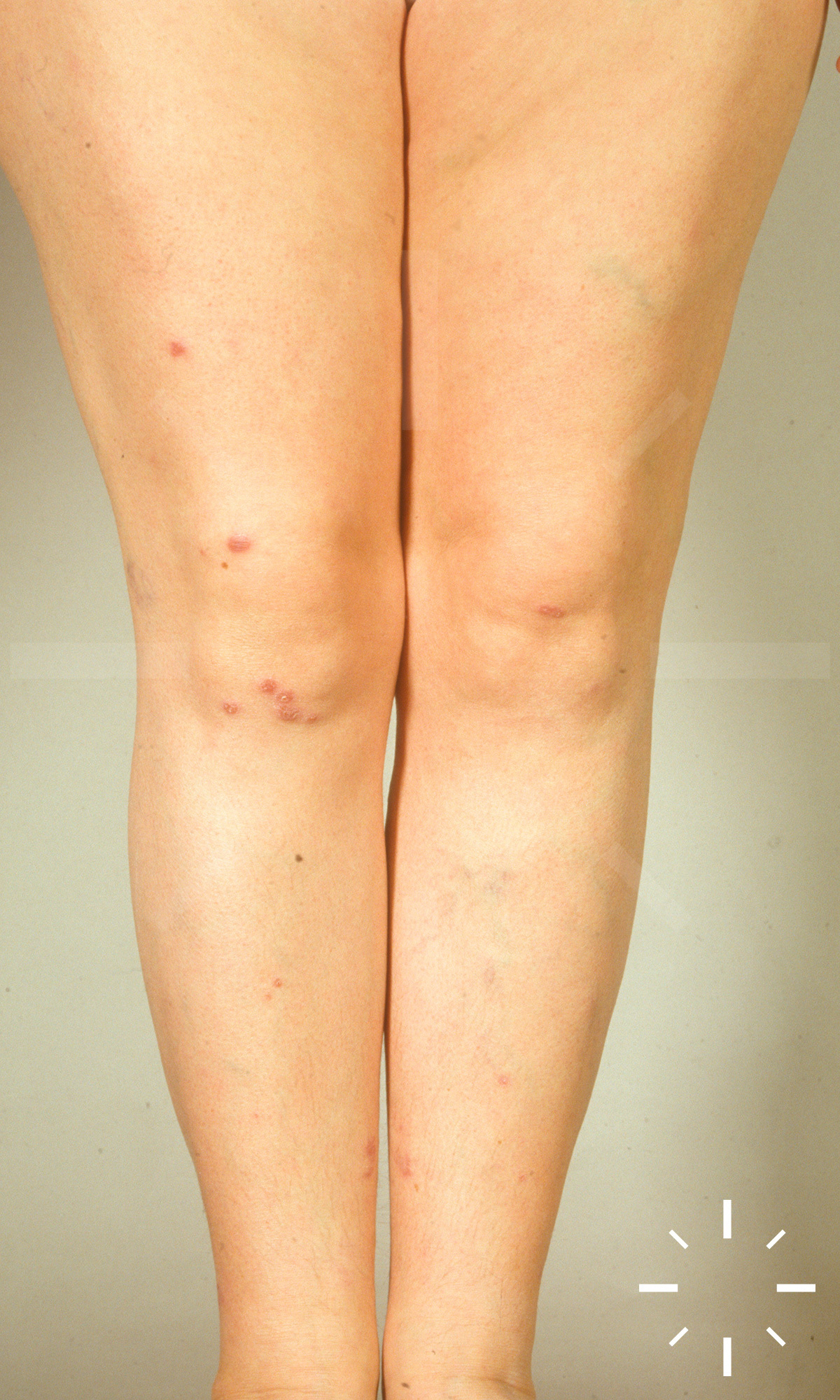
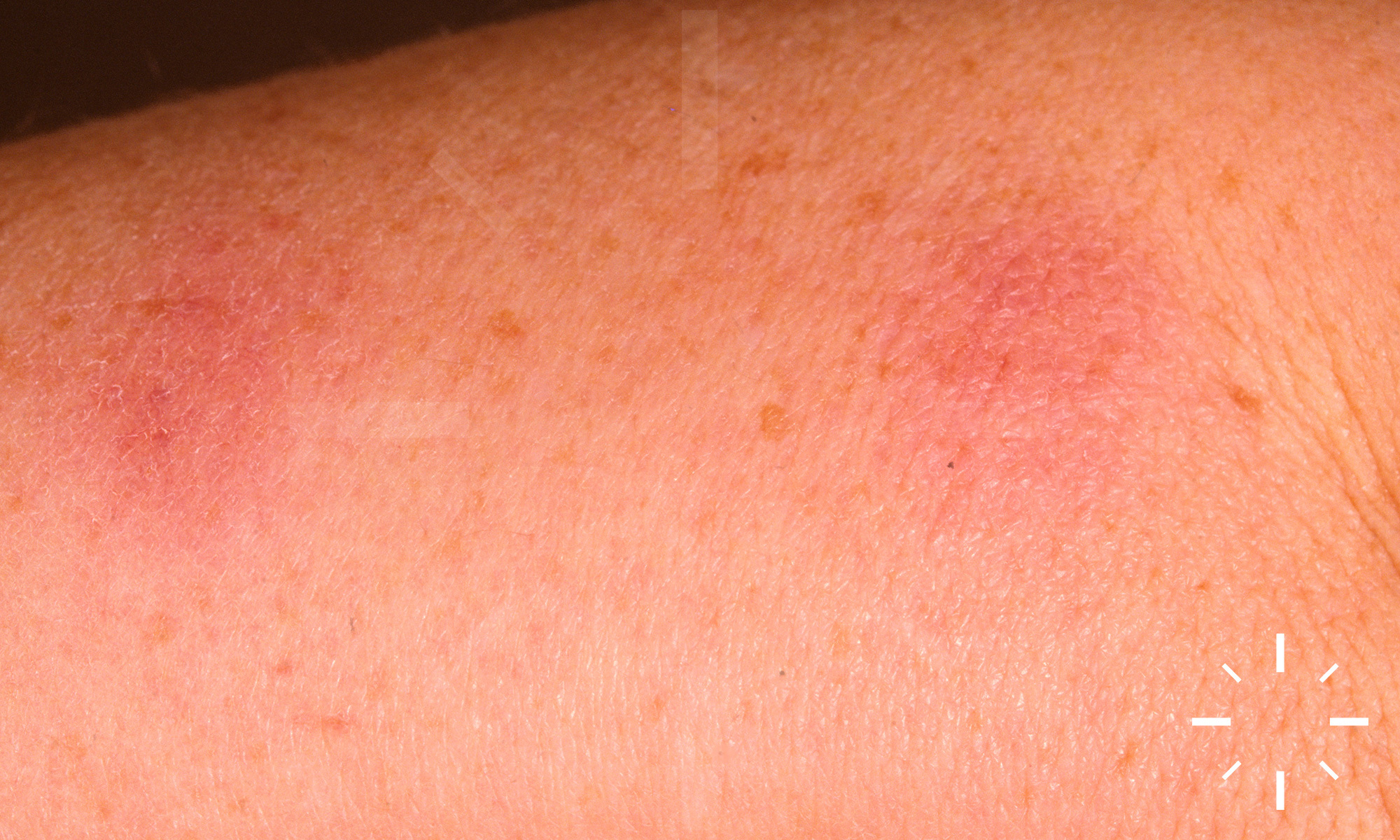
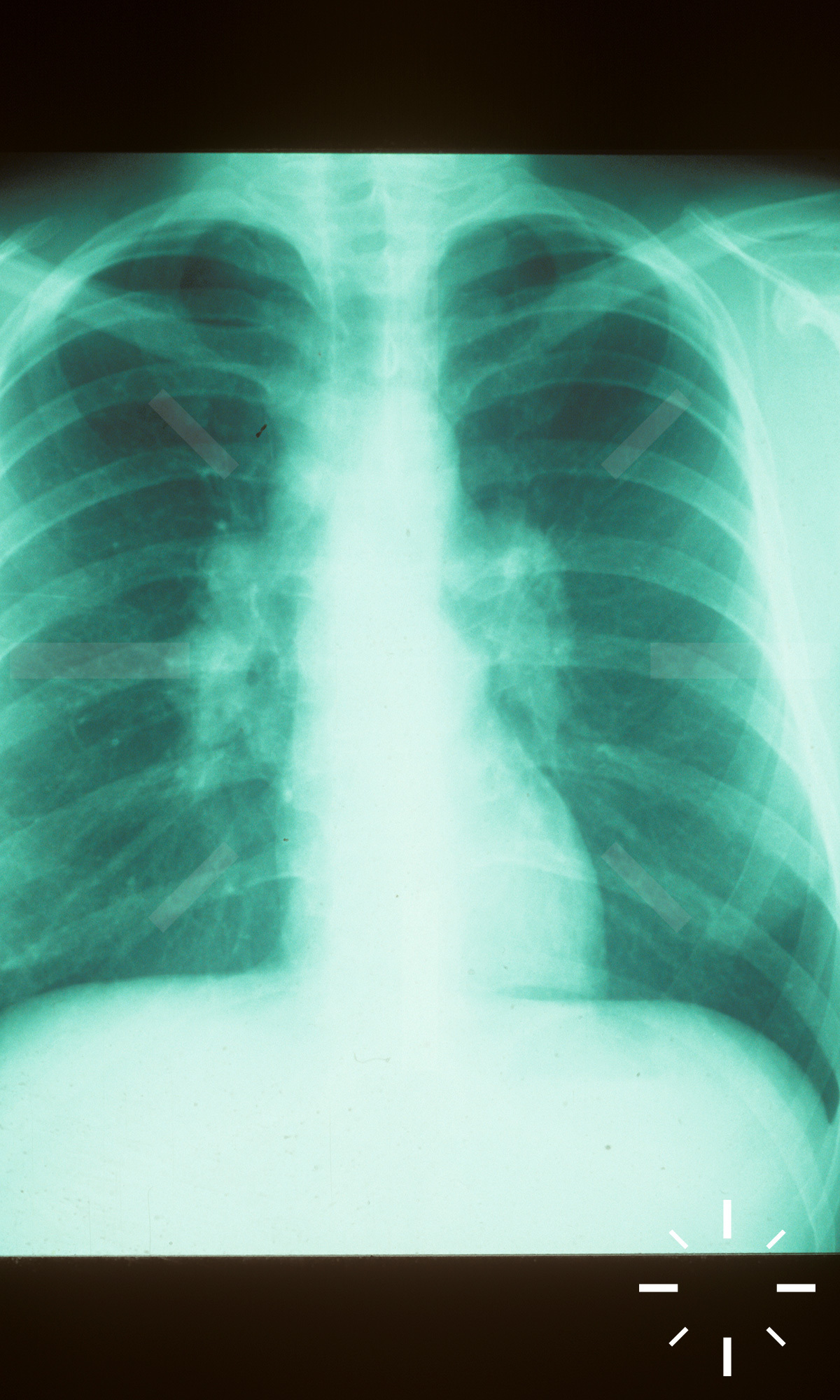
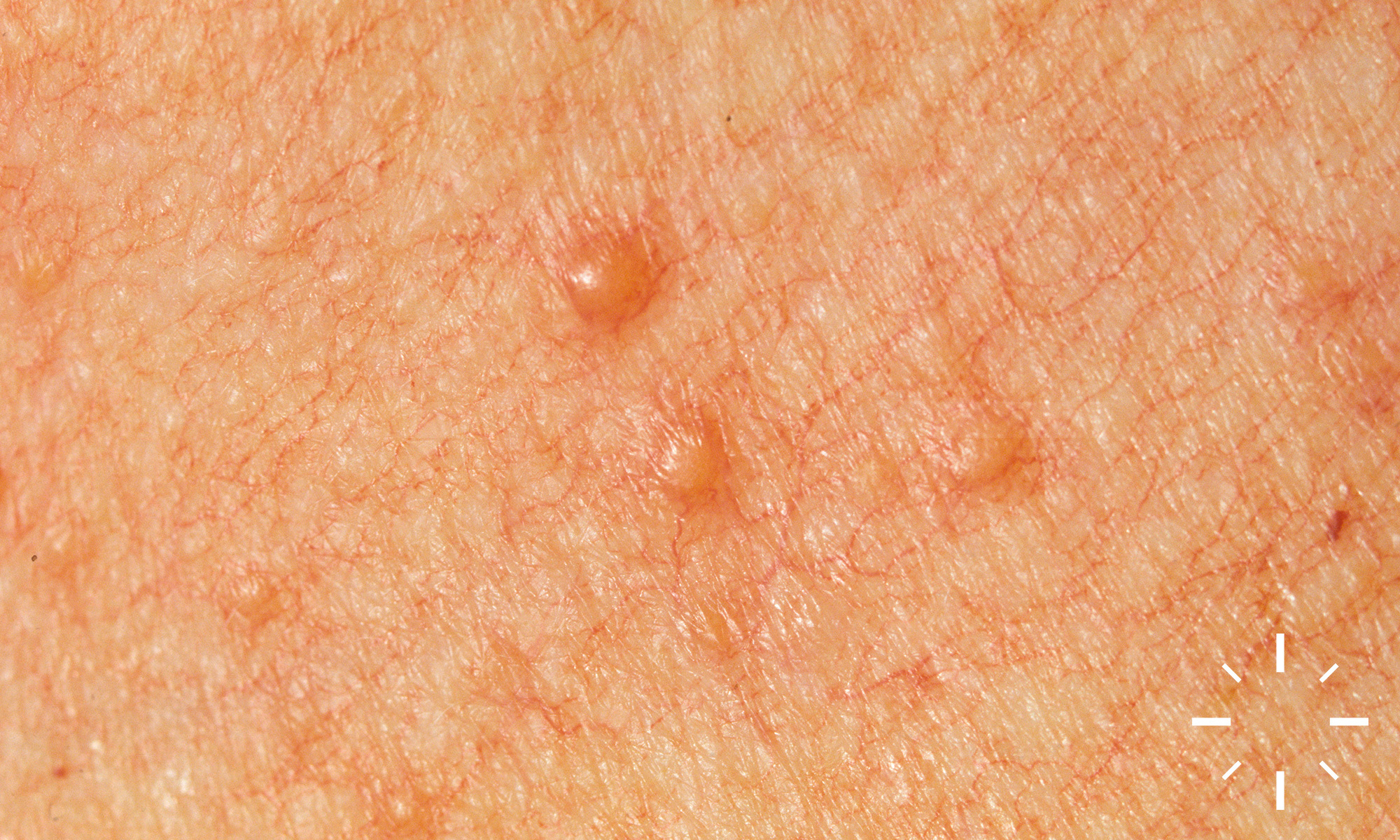
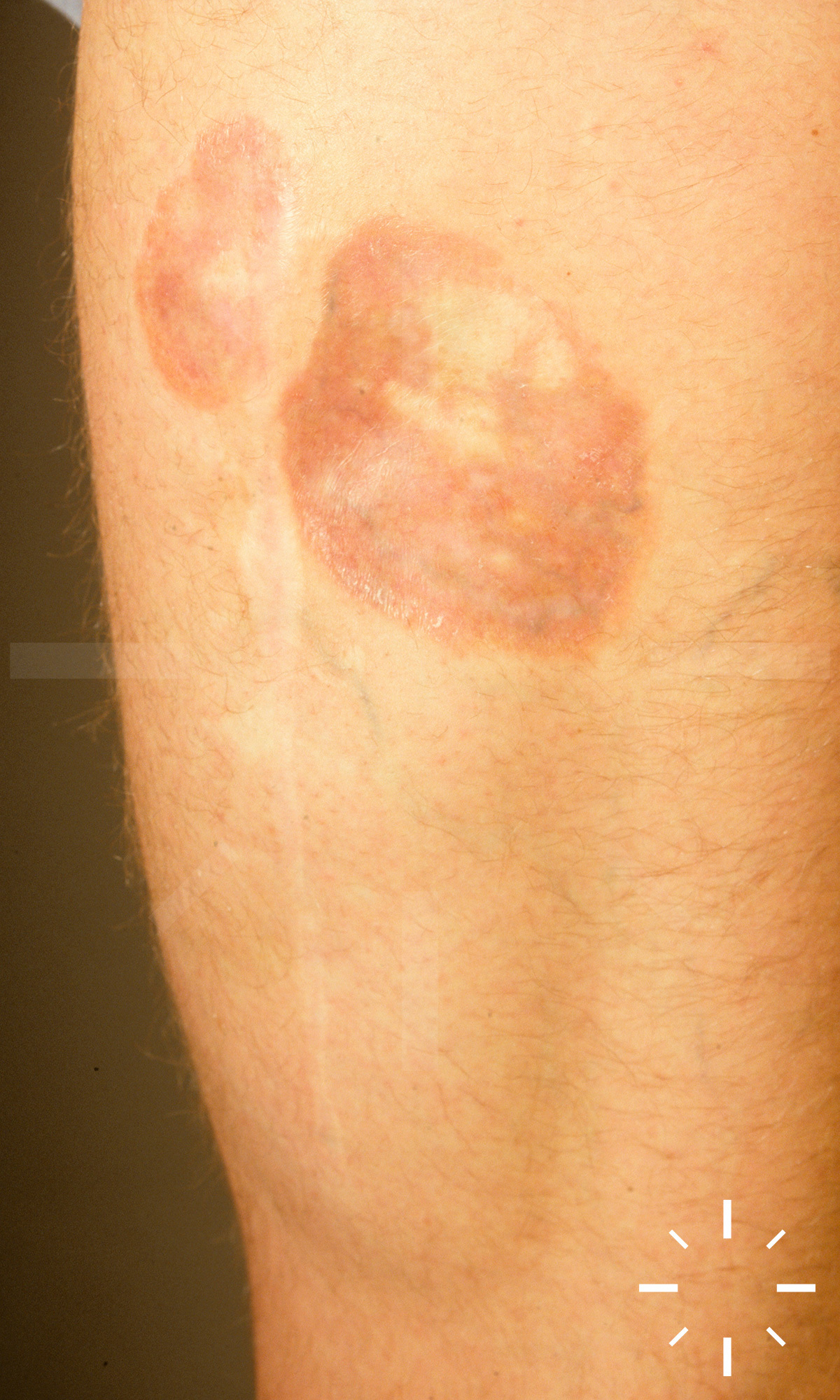
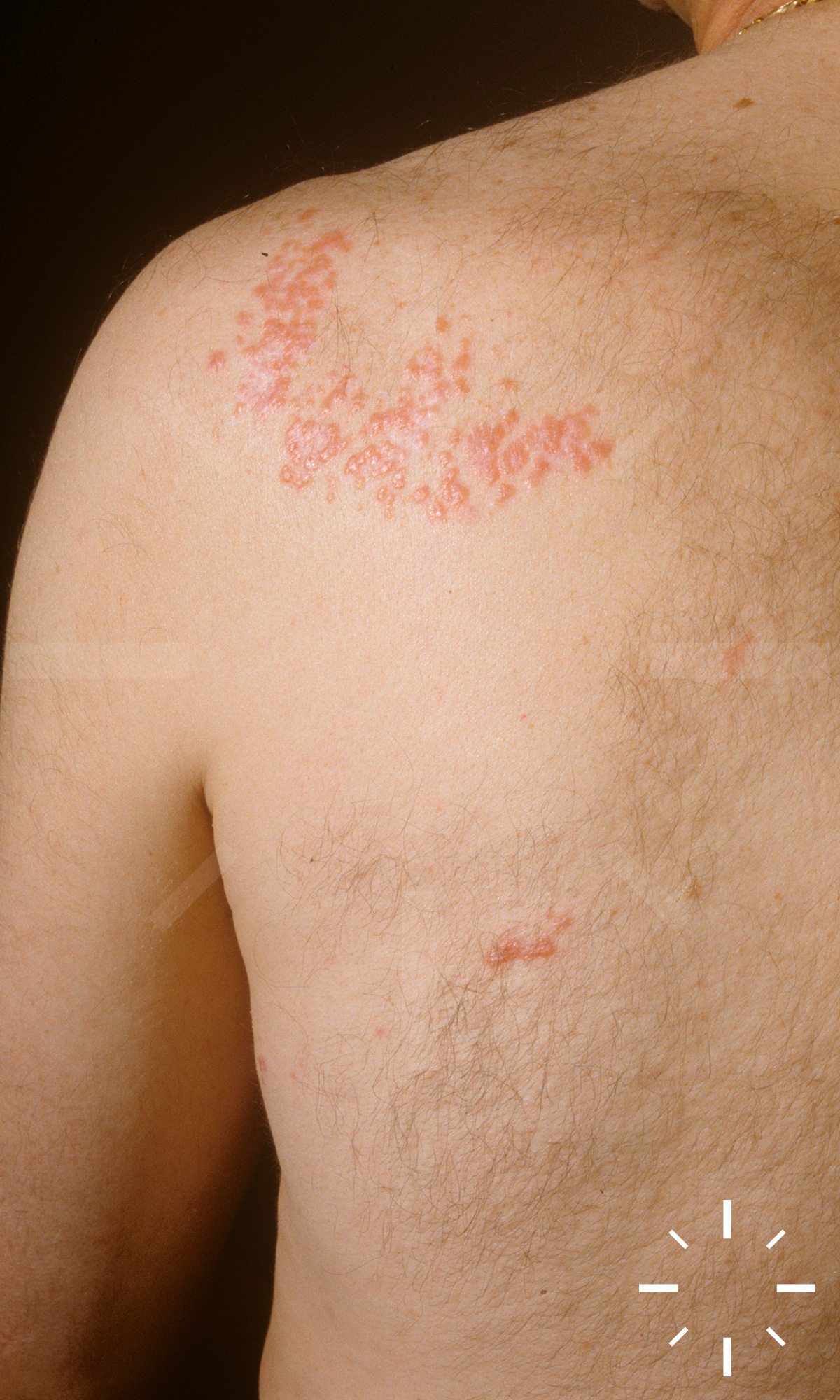
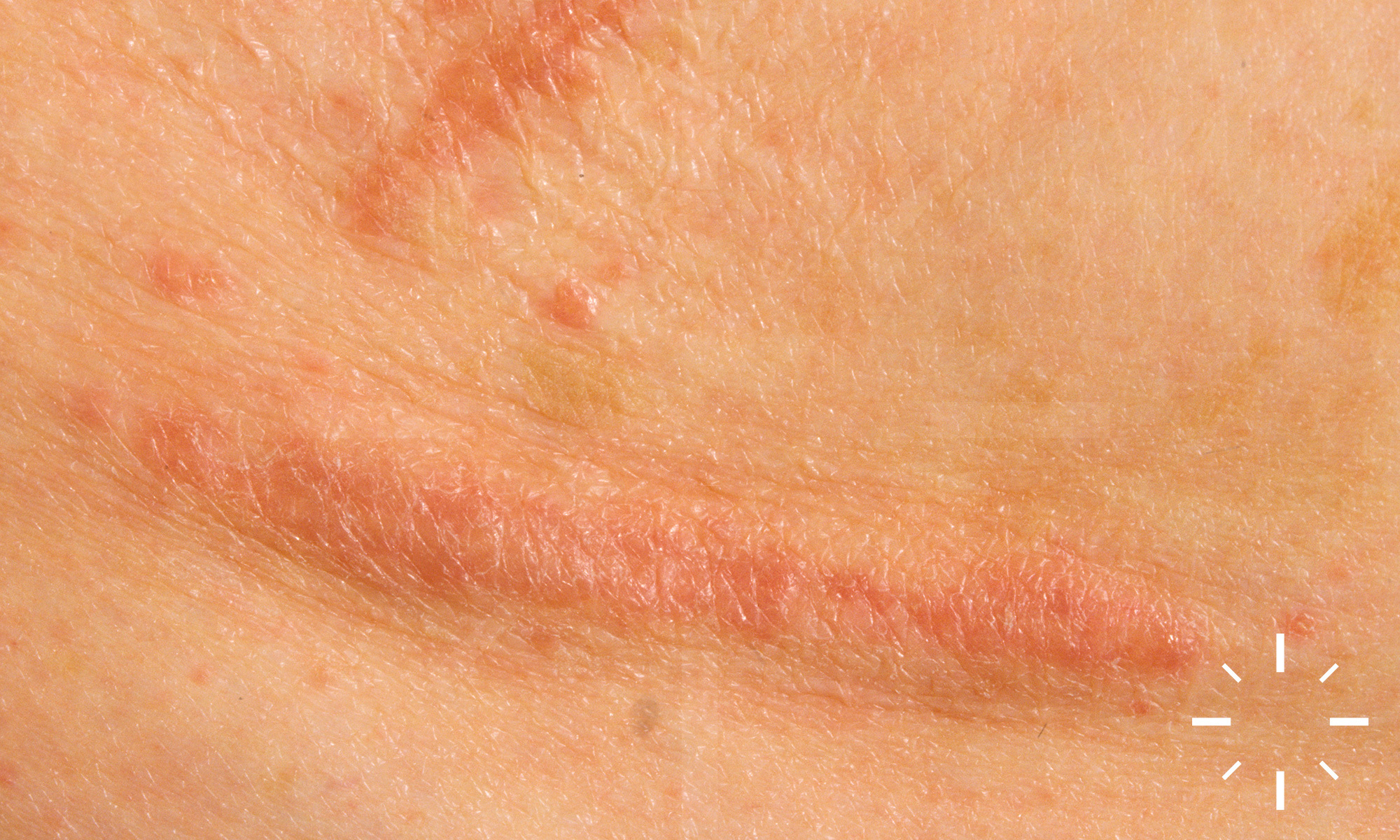
Sarkoidose
Zuletzt aktualisiert: 2024-10-29
Autor(en): Anzengruber F., Navarini A.
ICD11: 4B20.Z
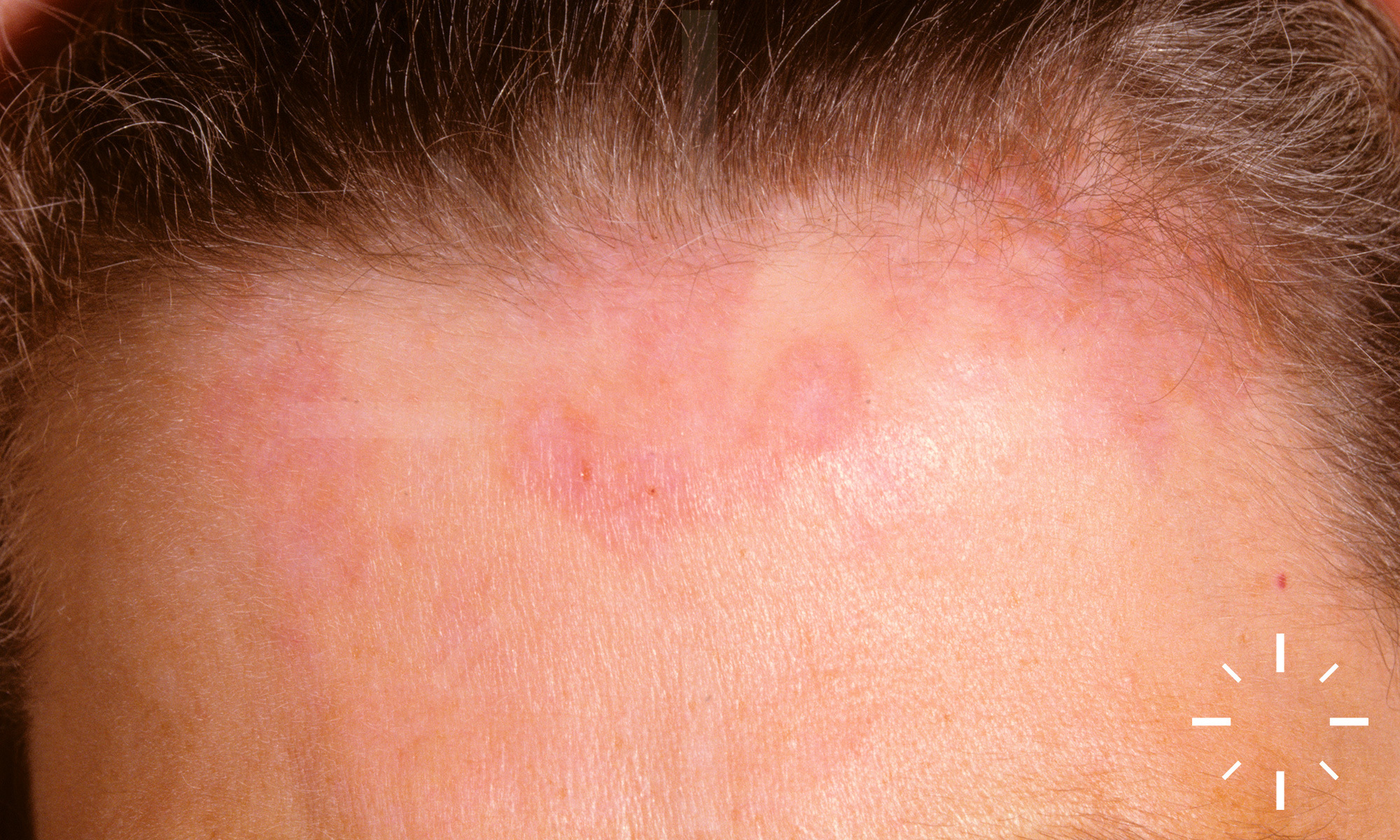
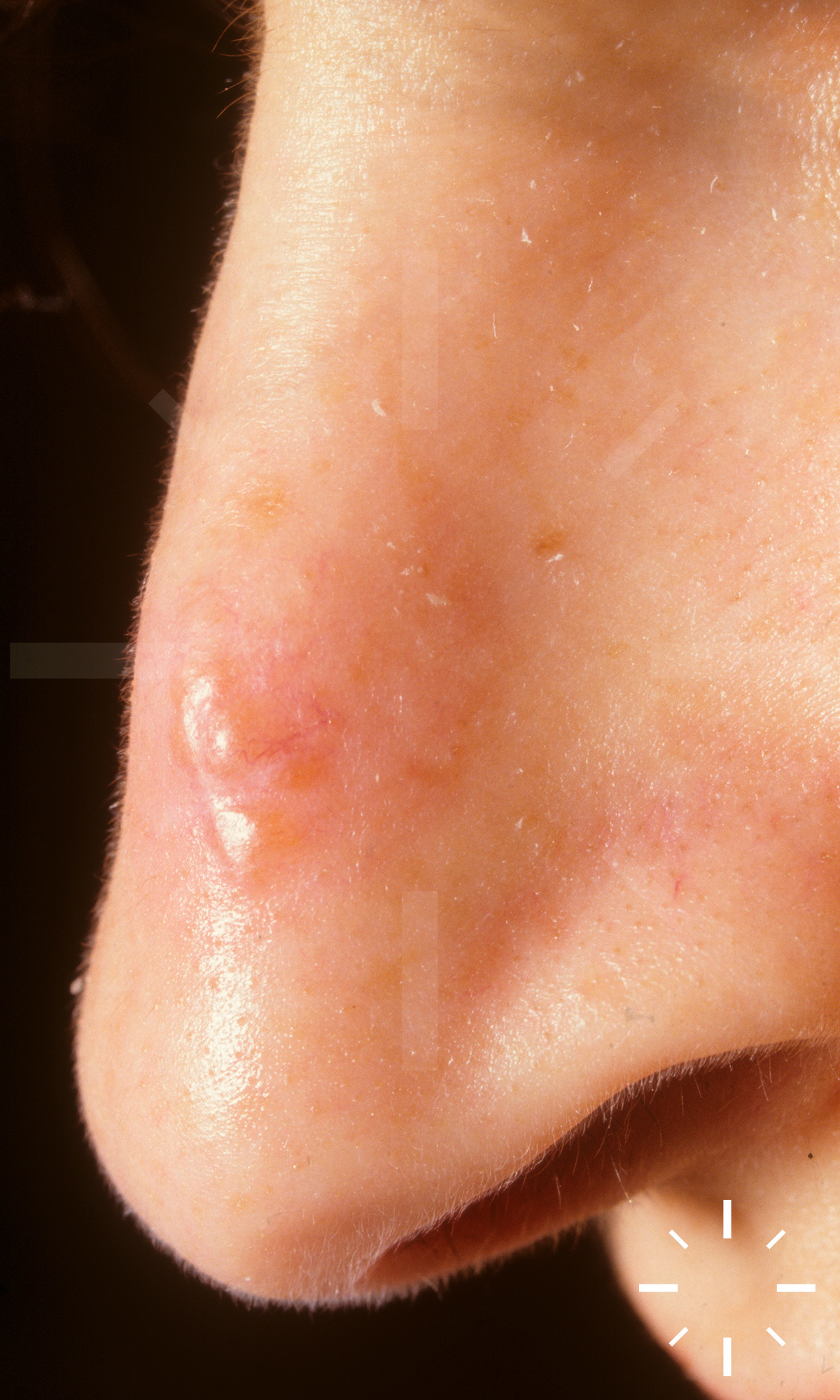
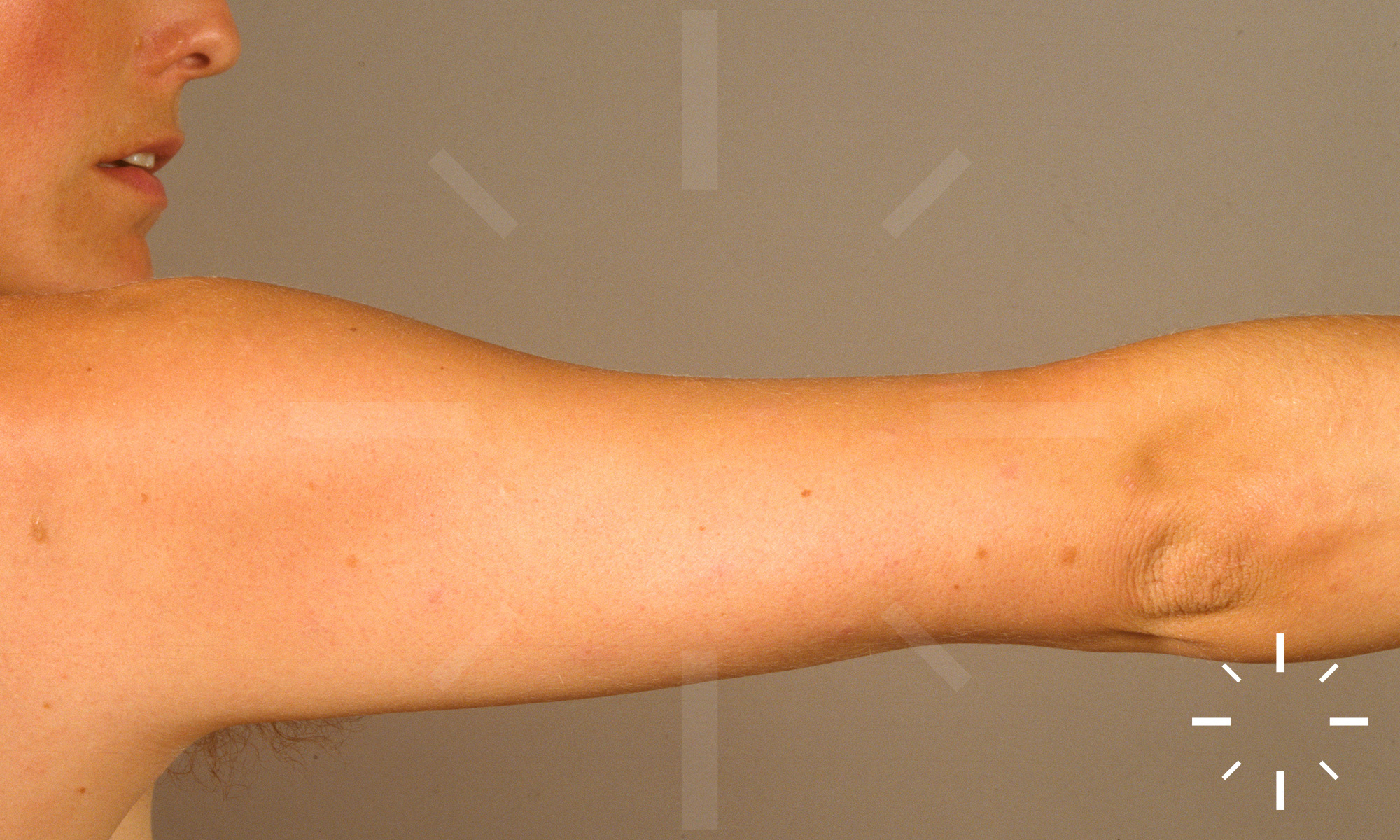
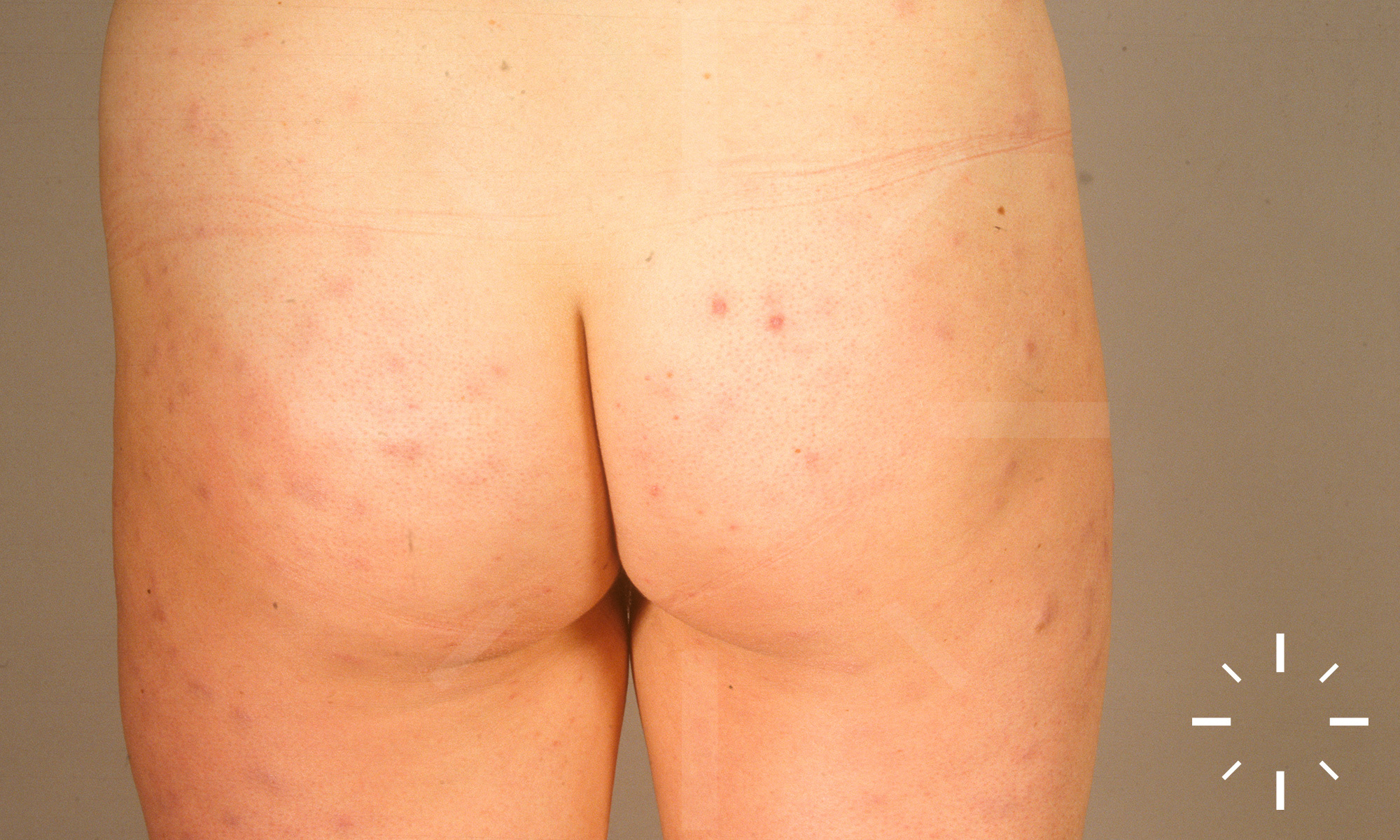
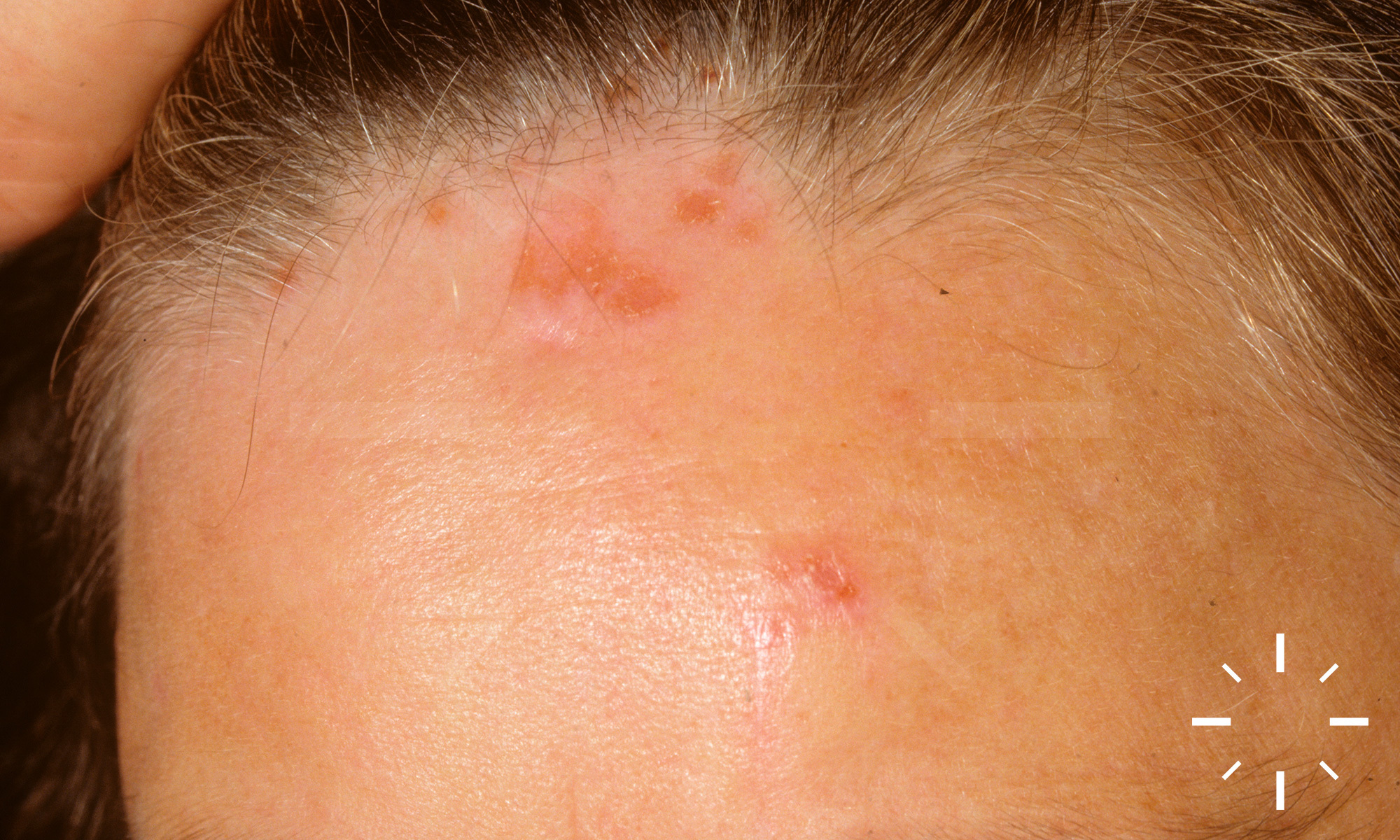
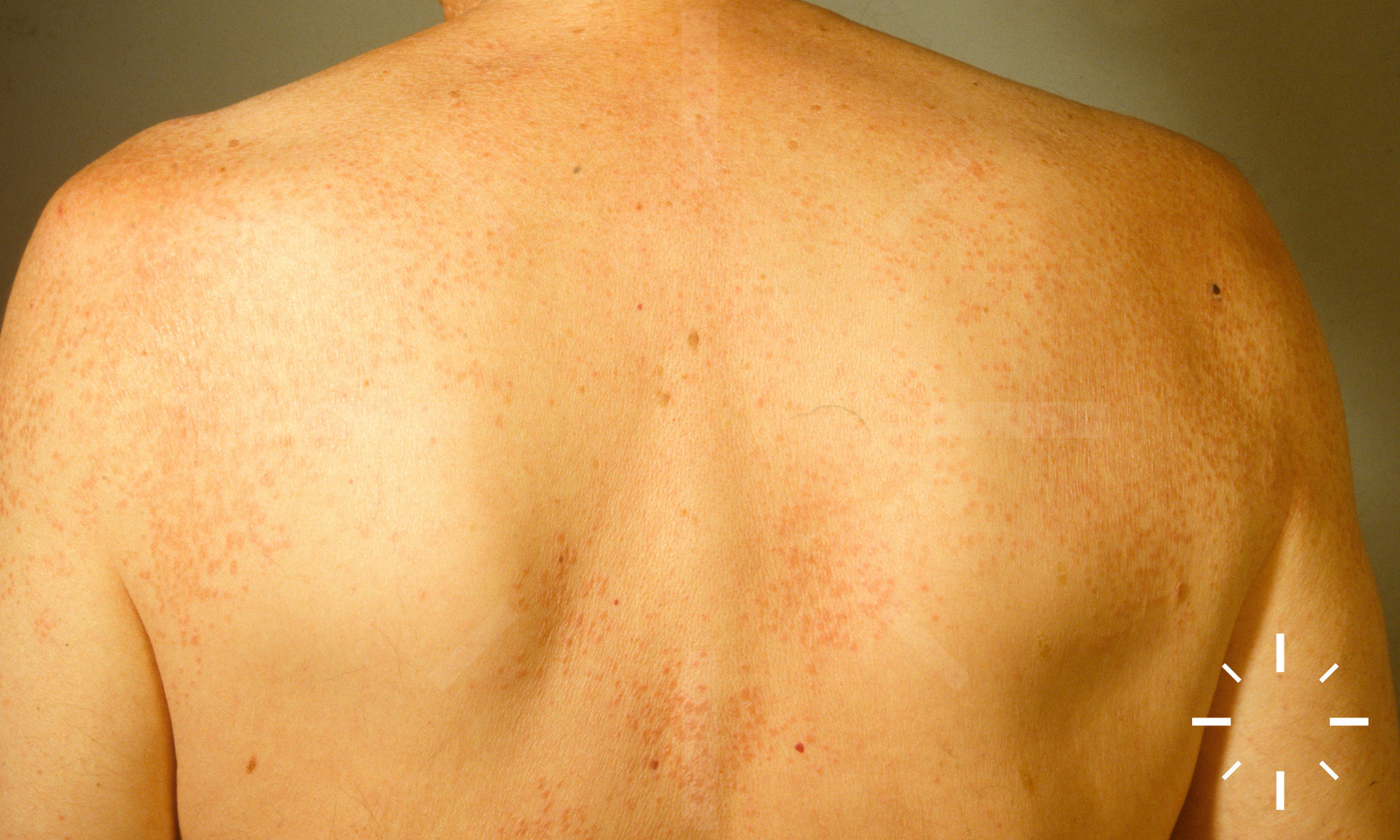
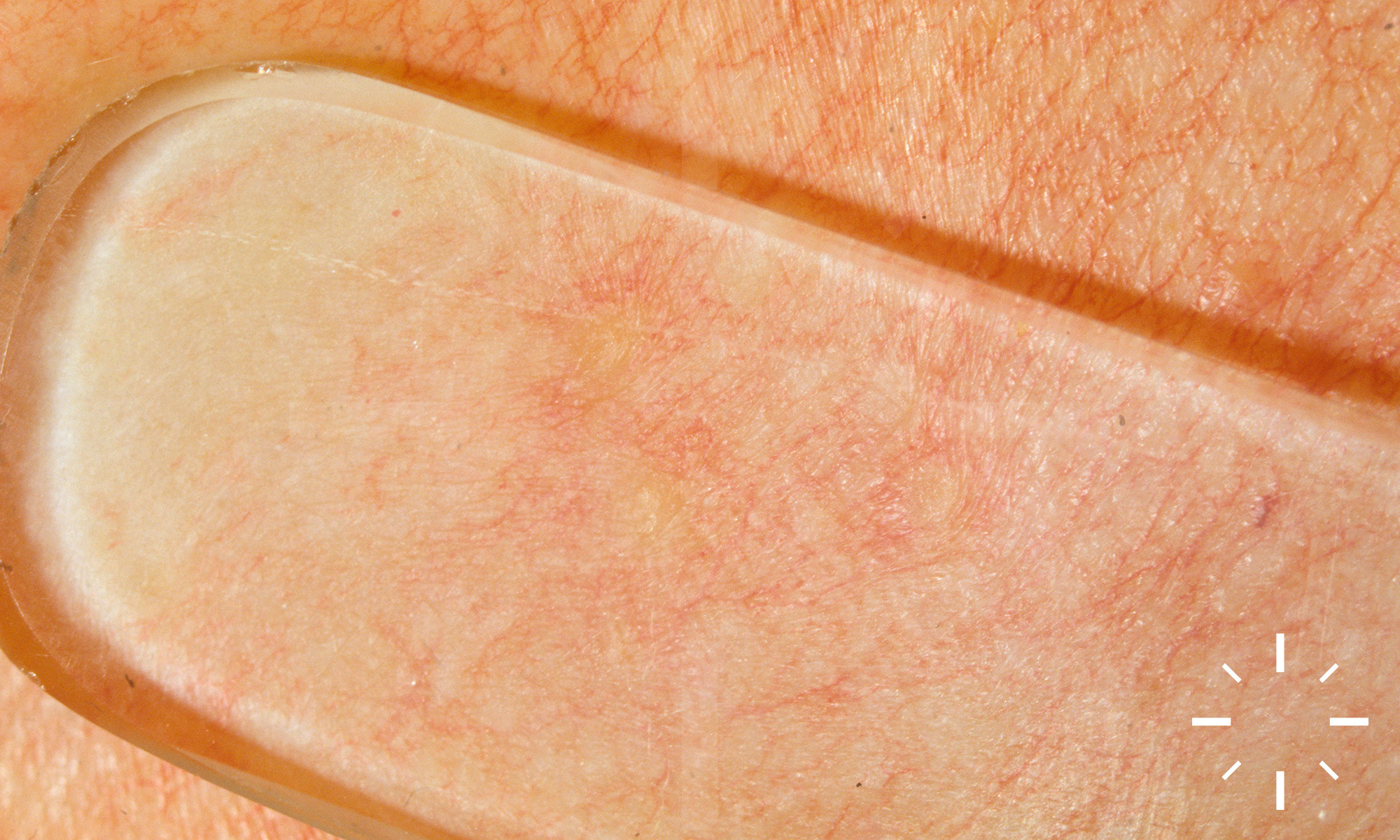
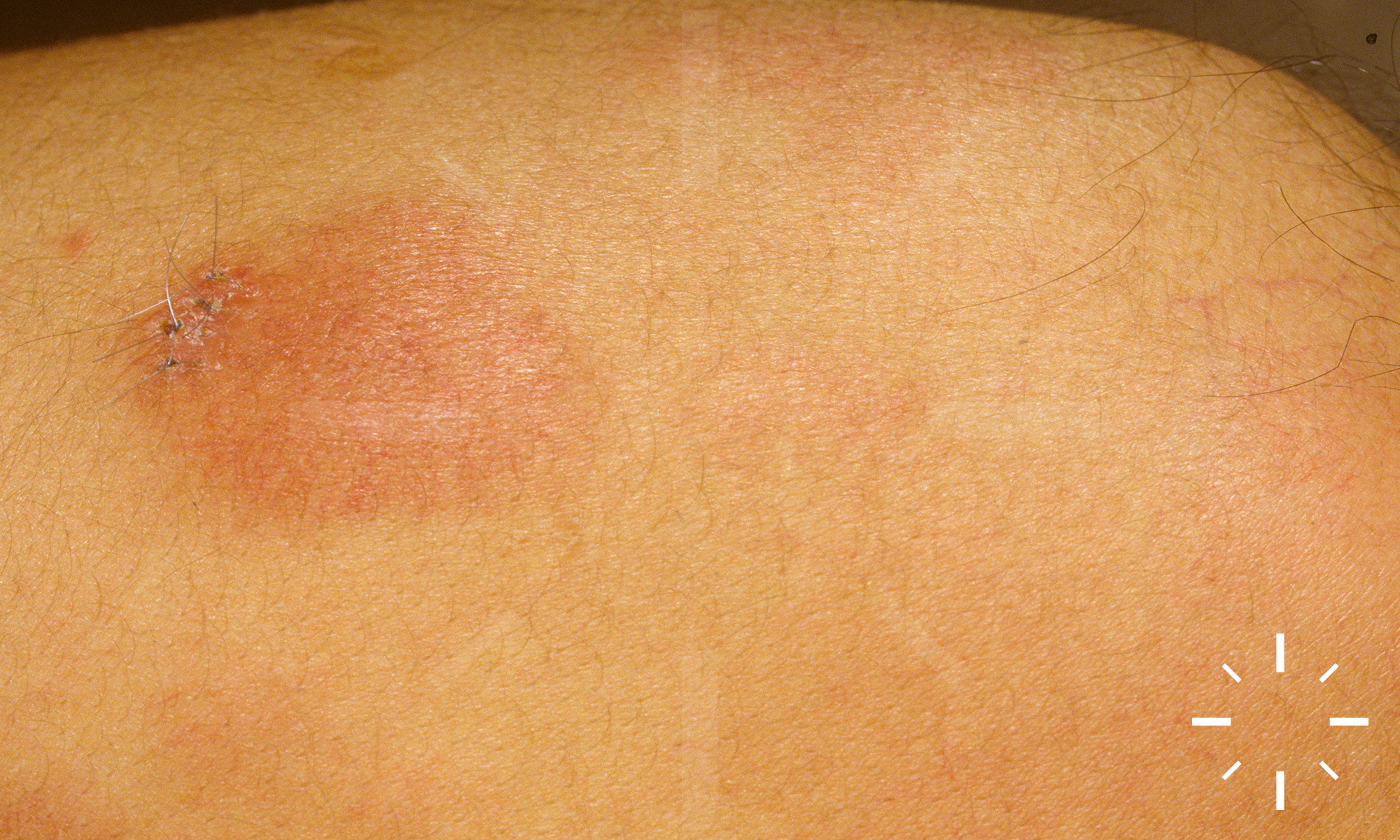
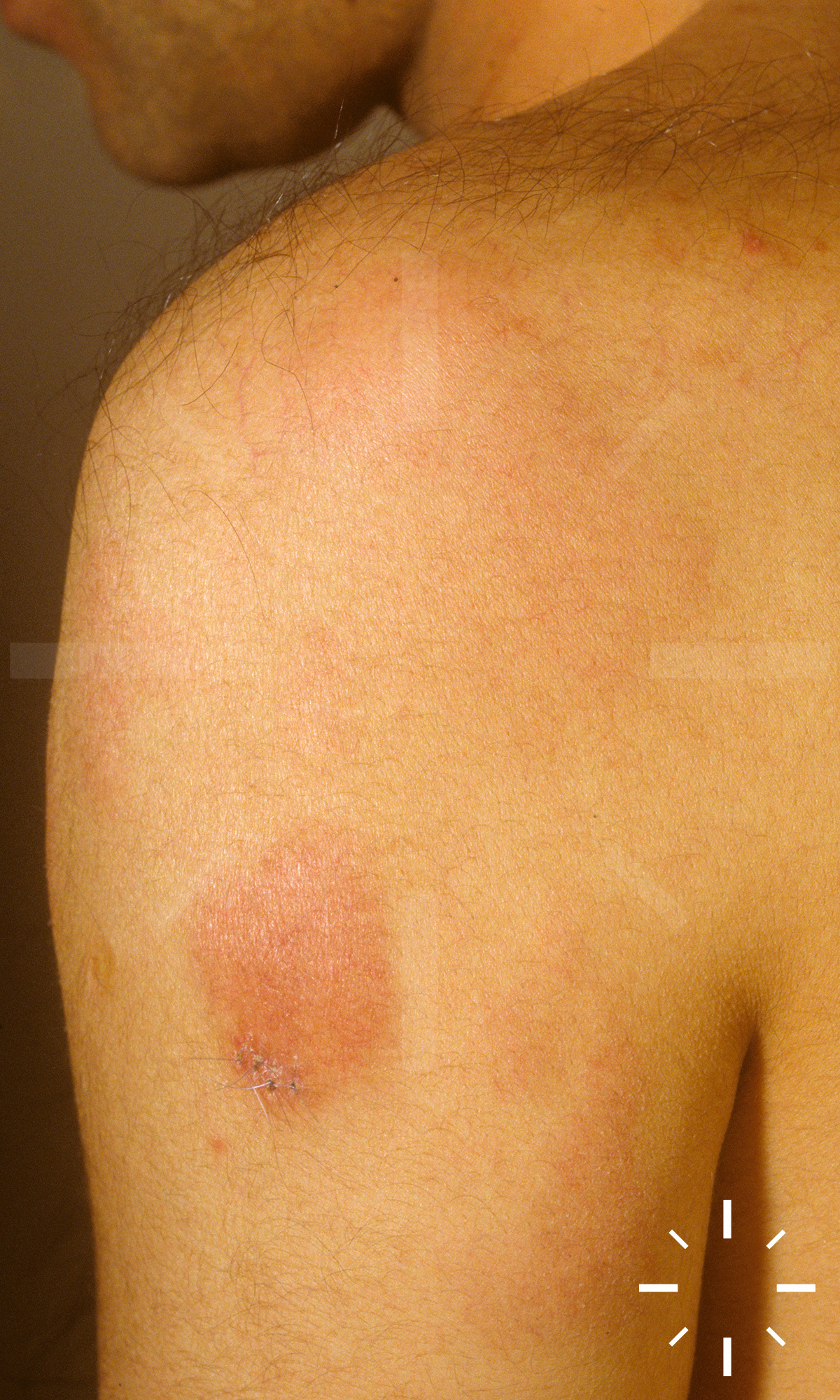
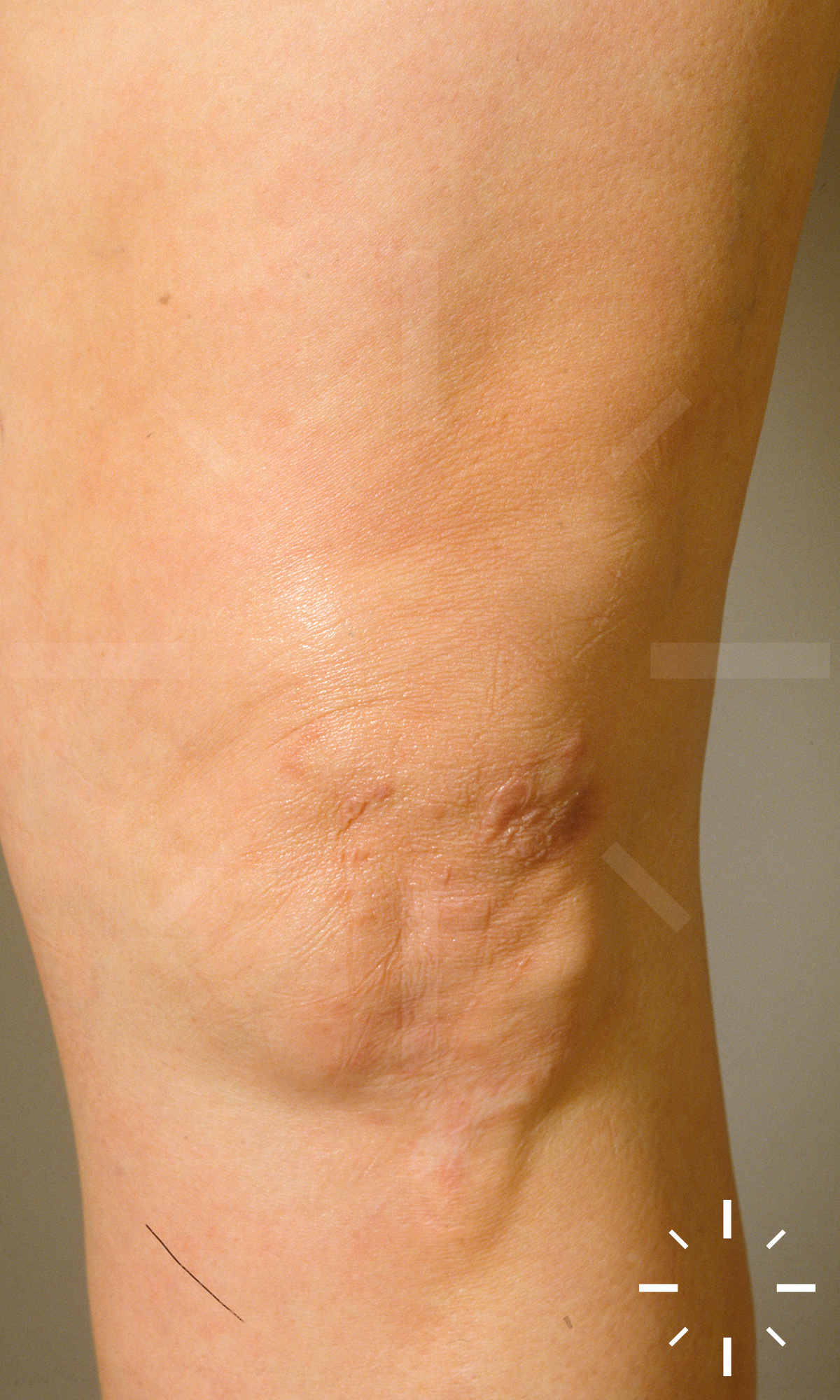
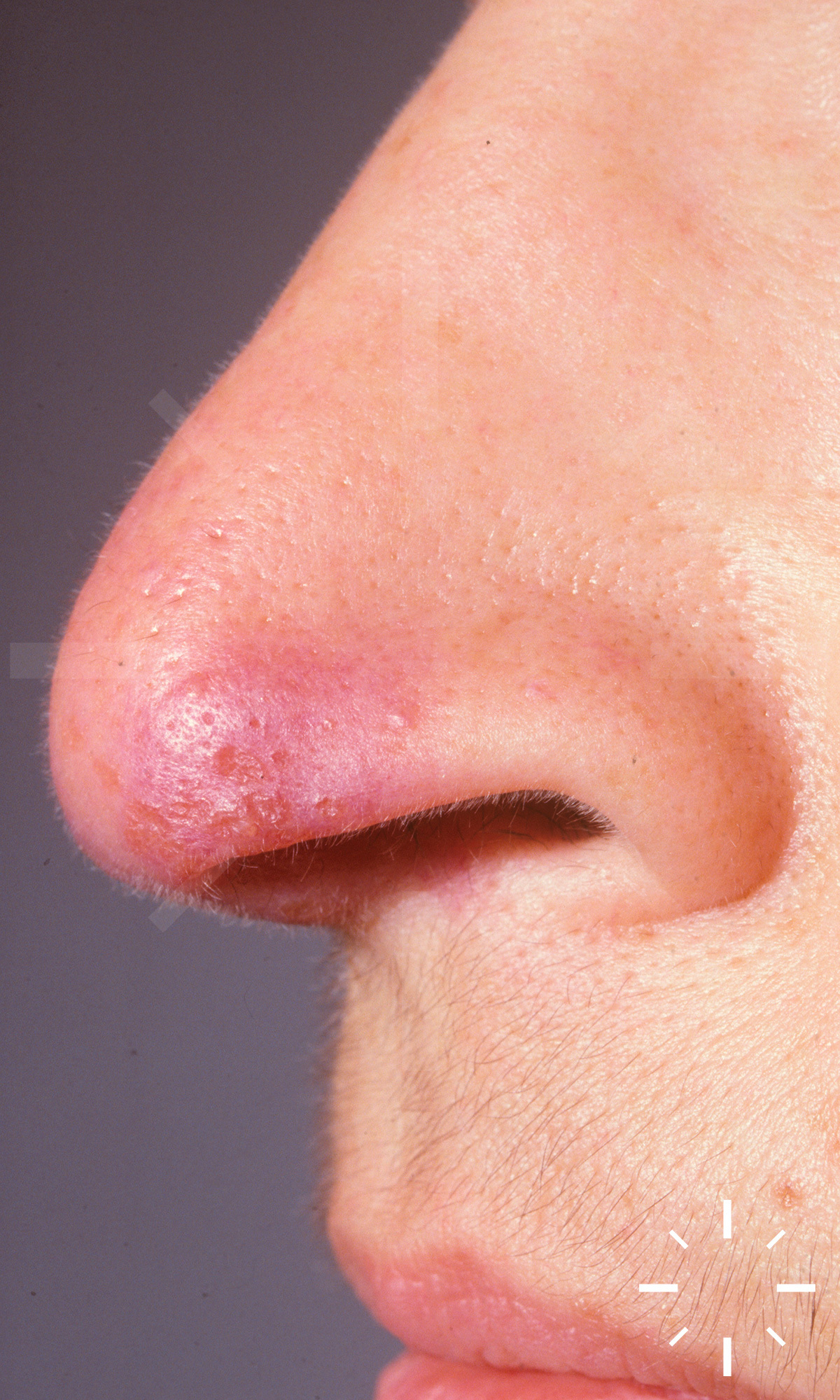
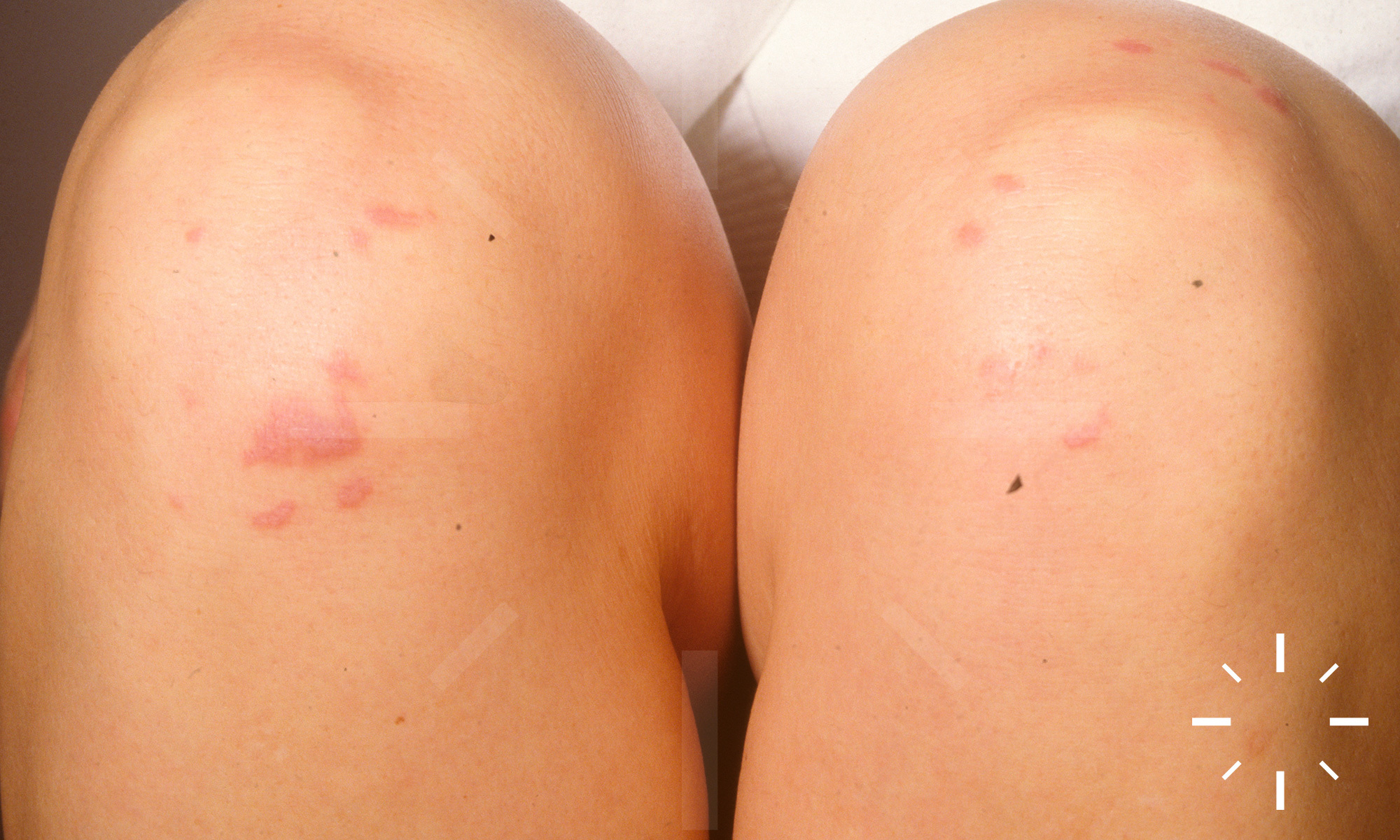
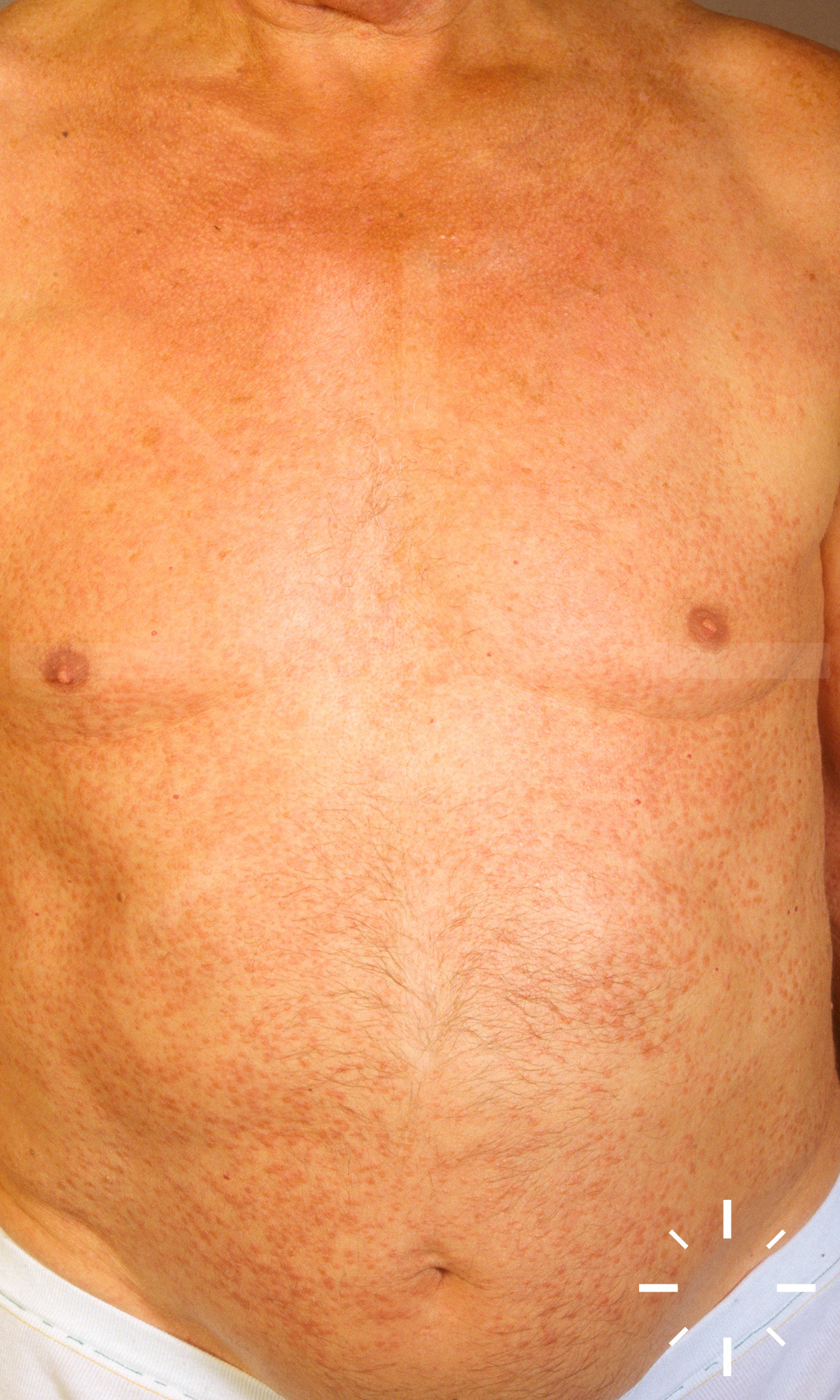
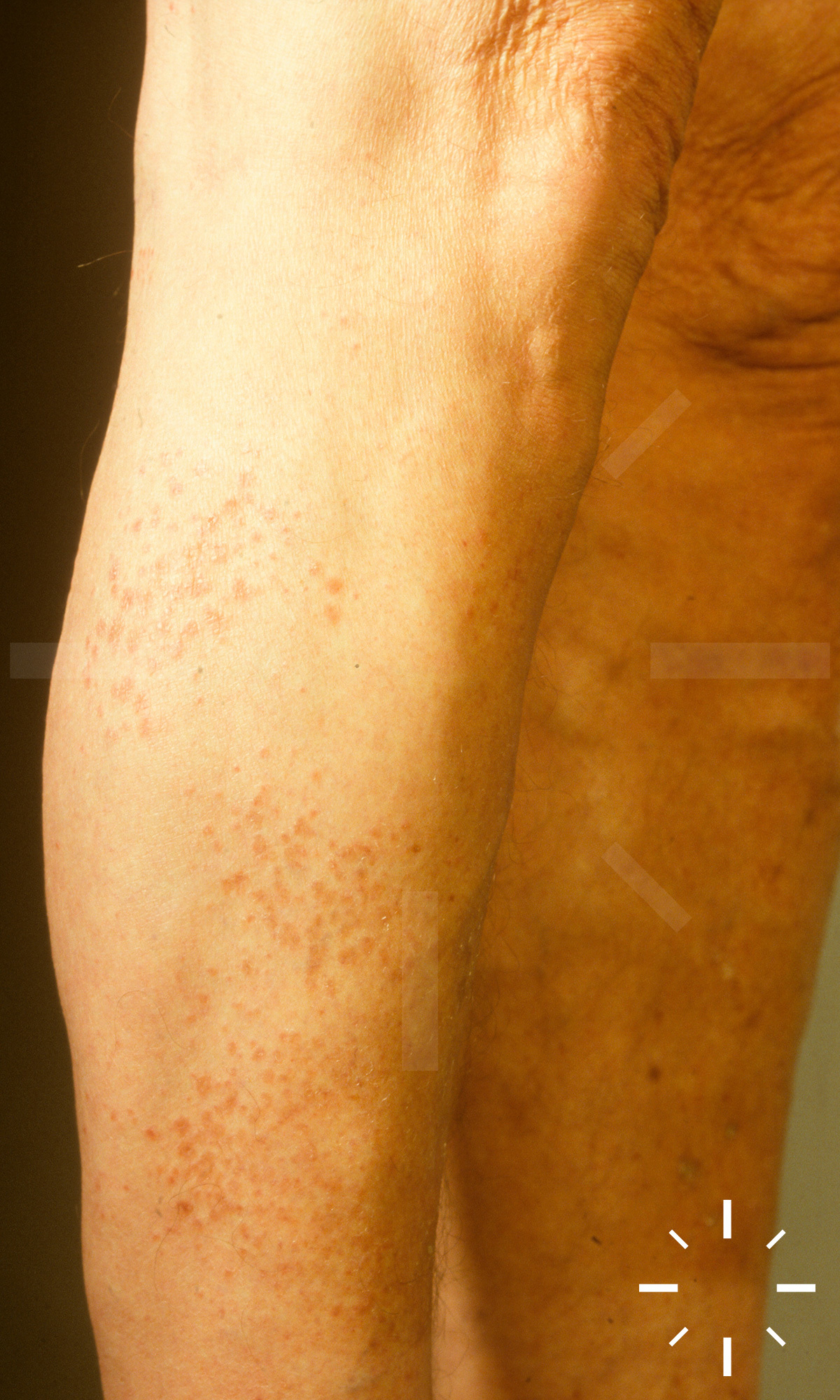
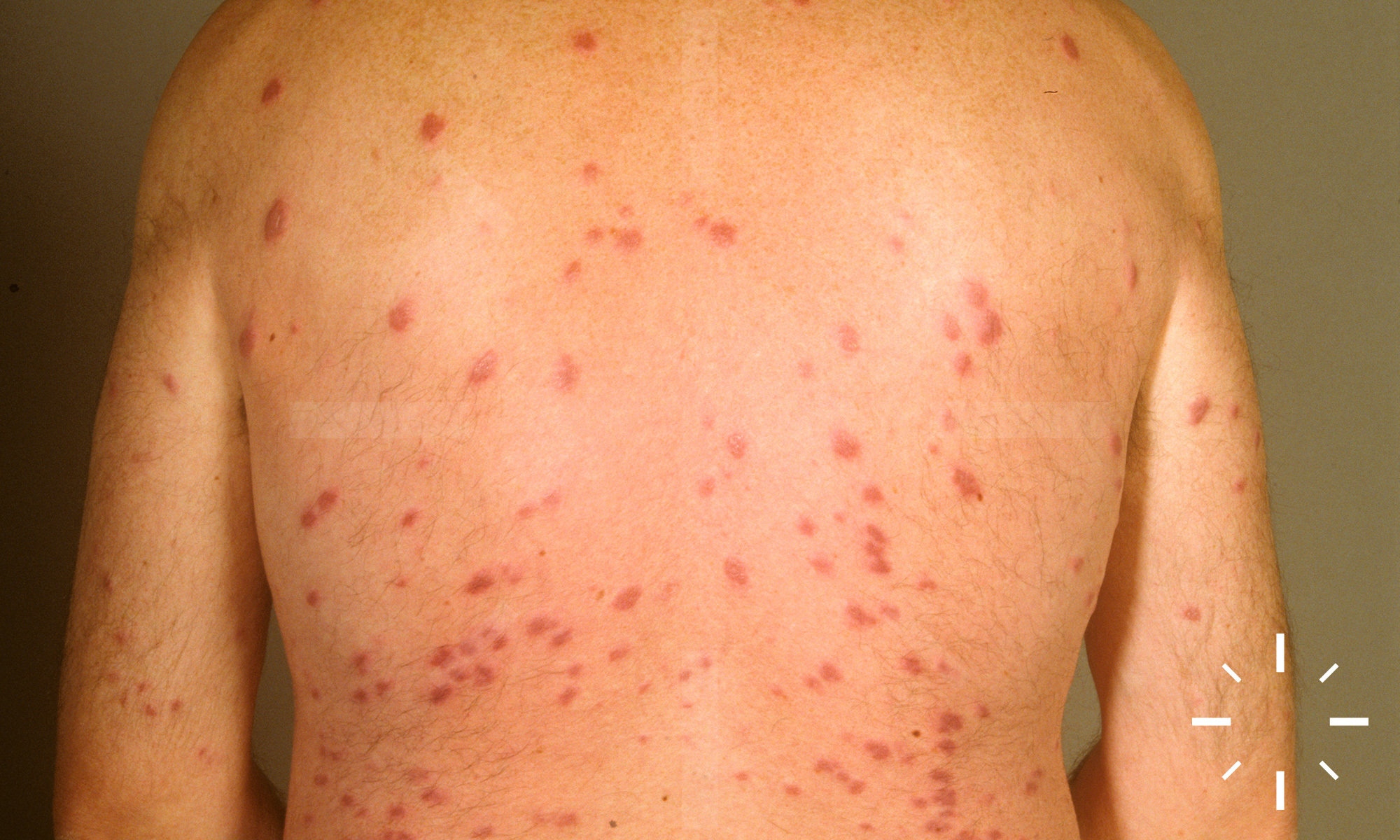
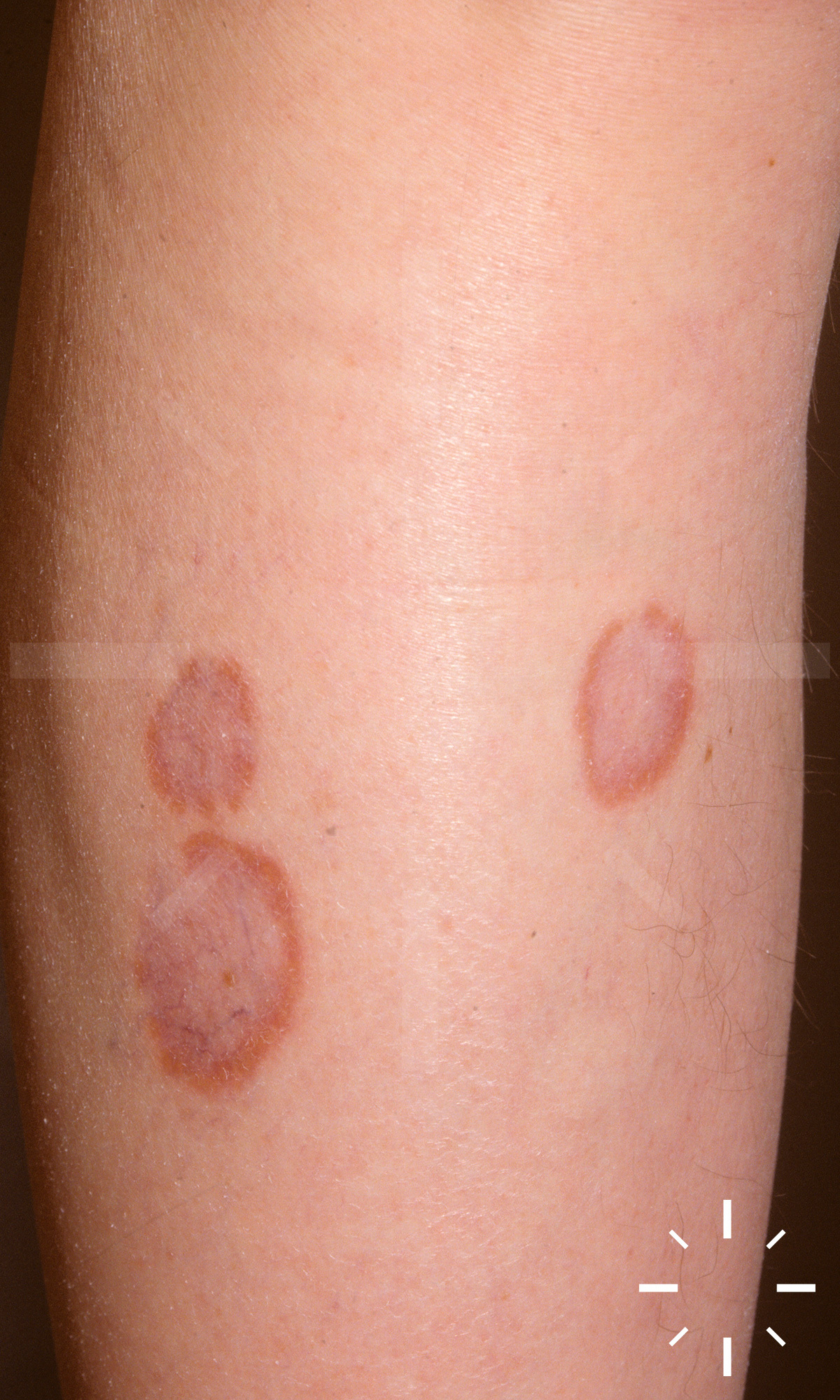


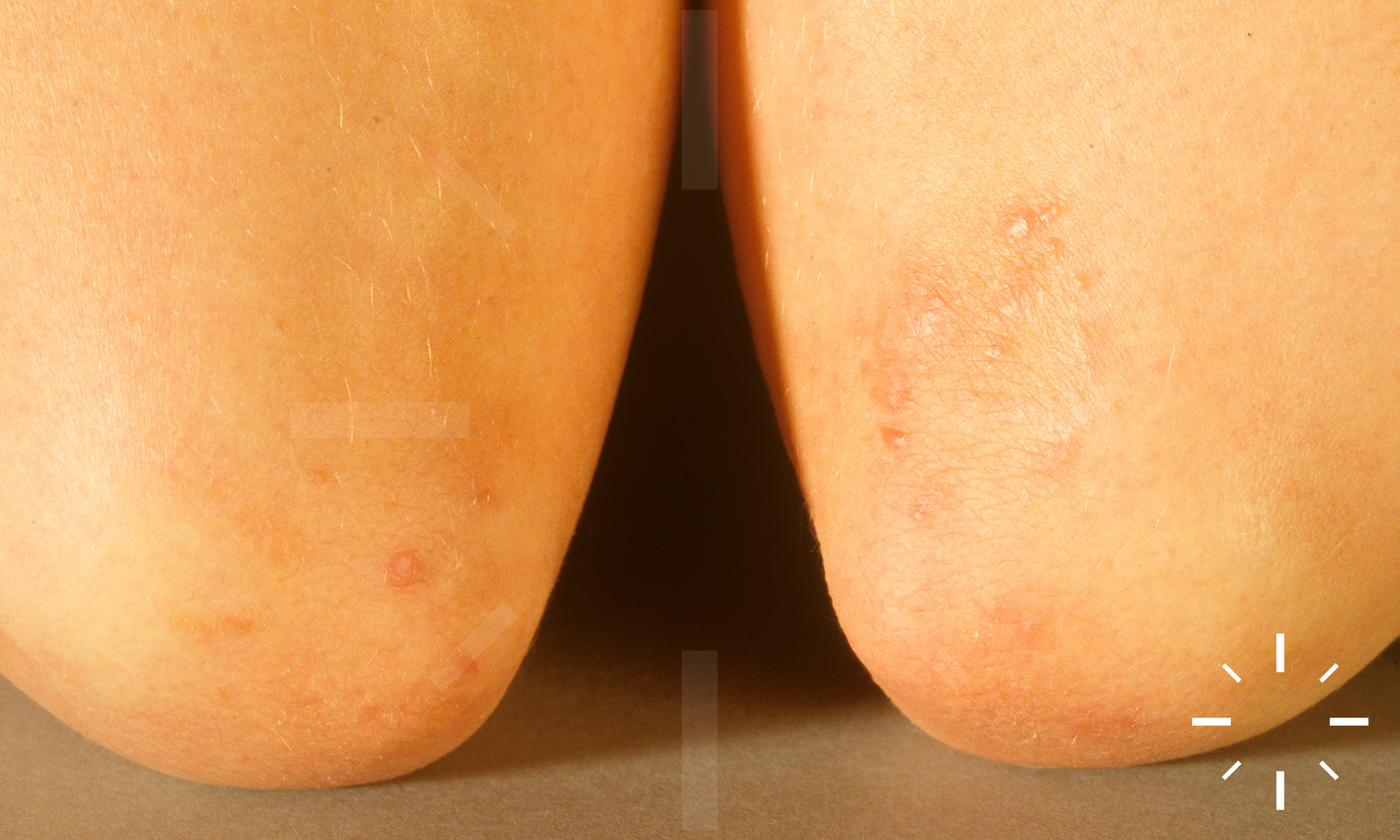
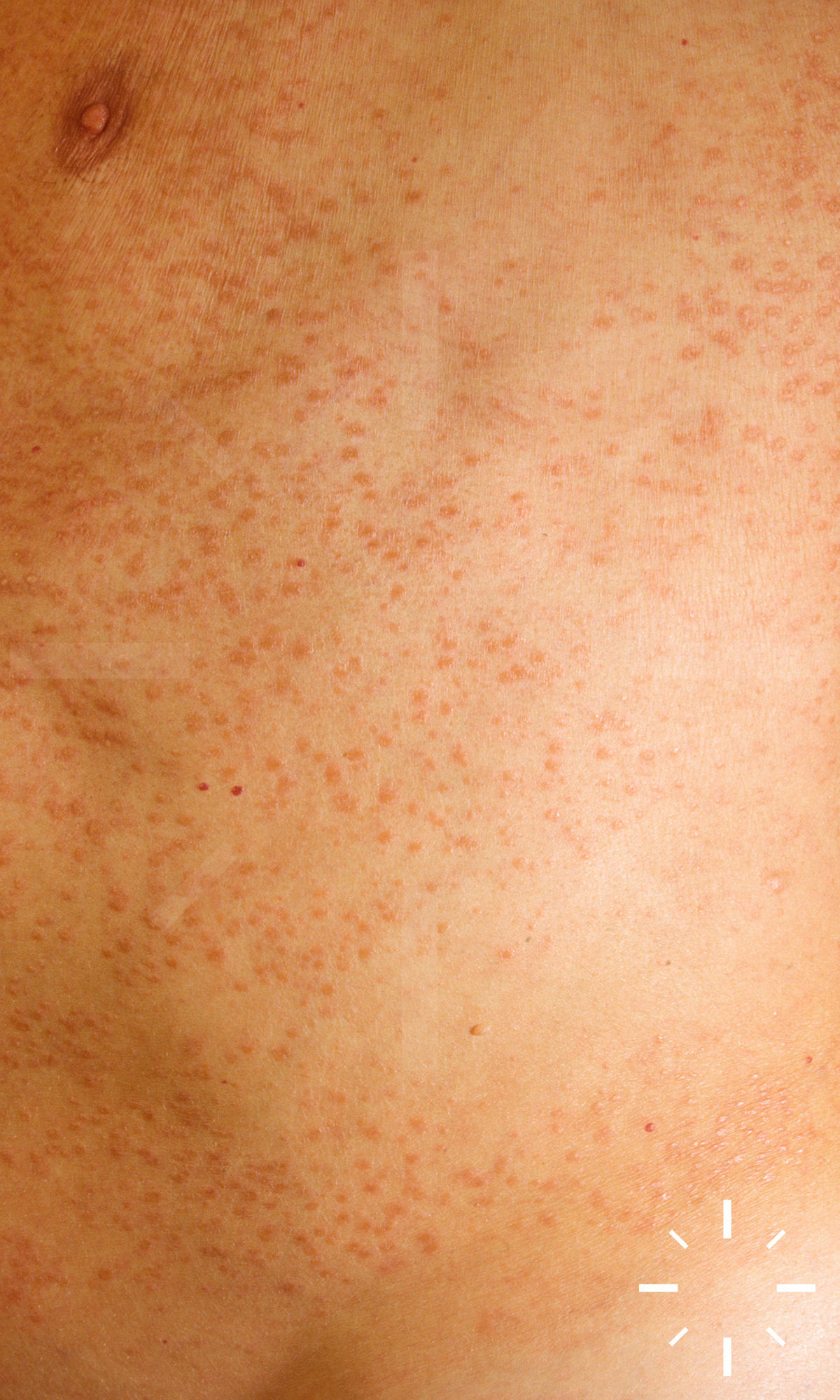
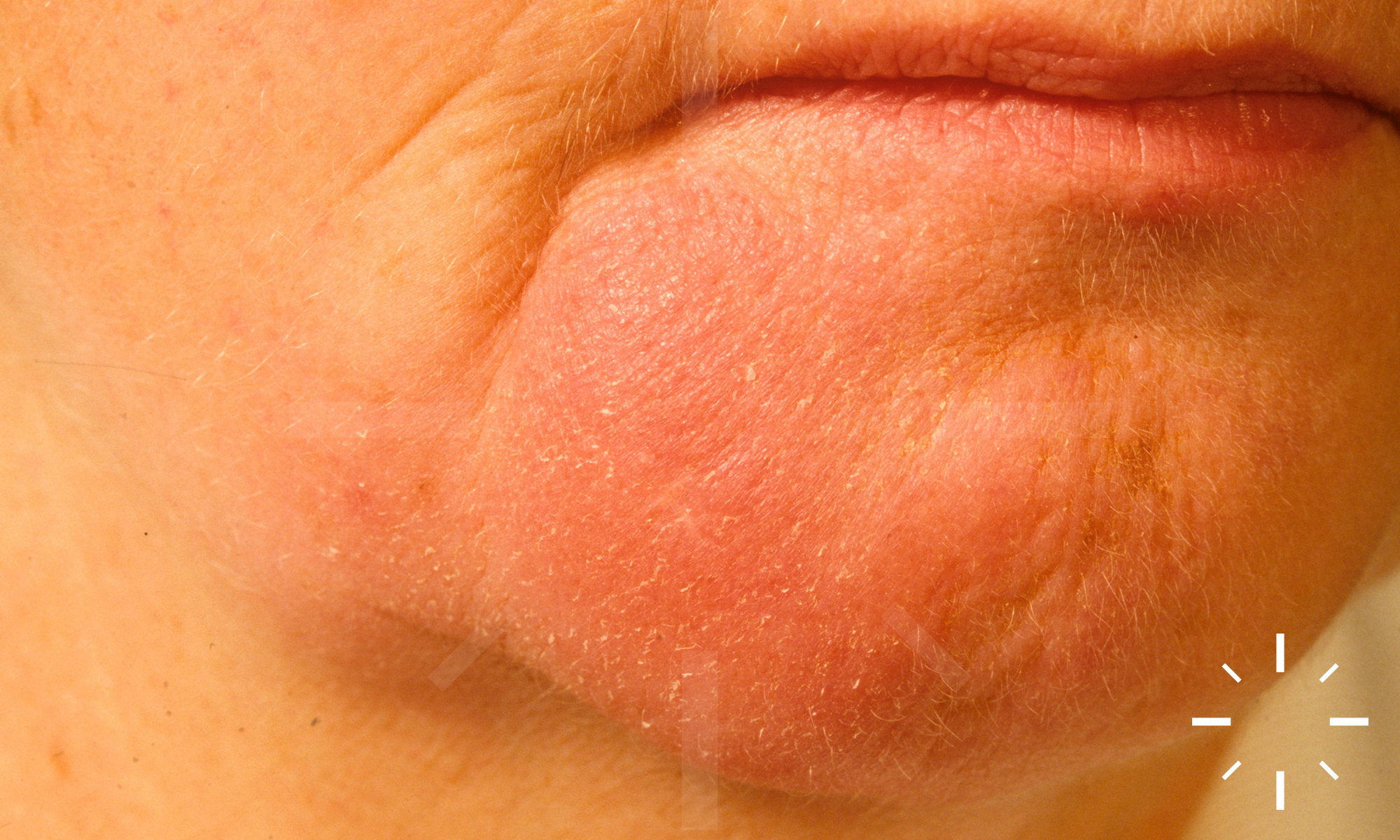
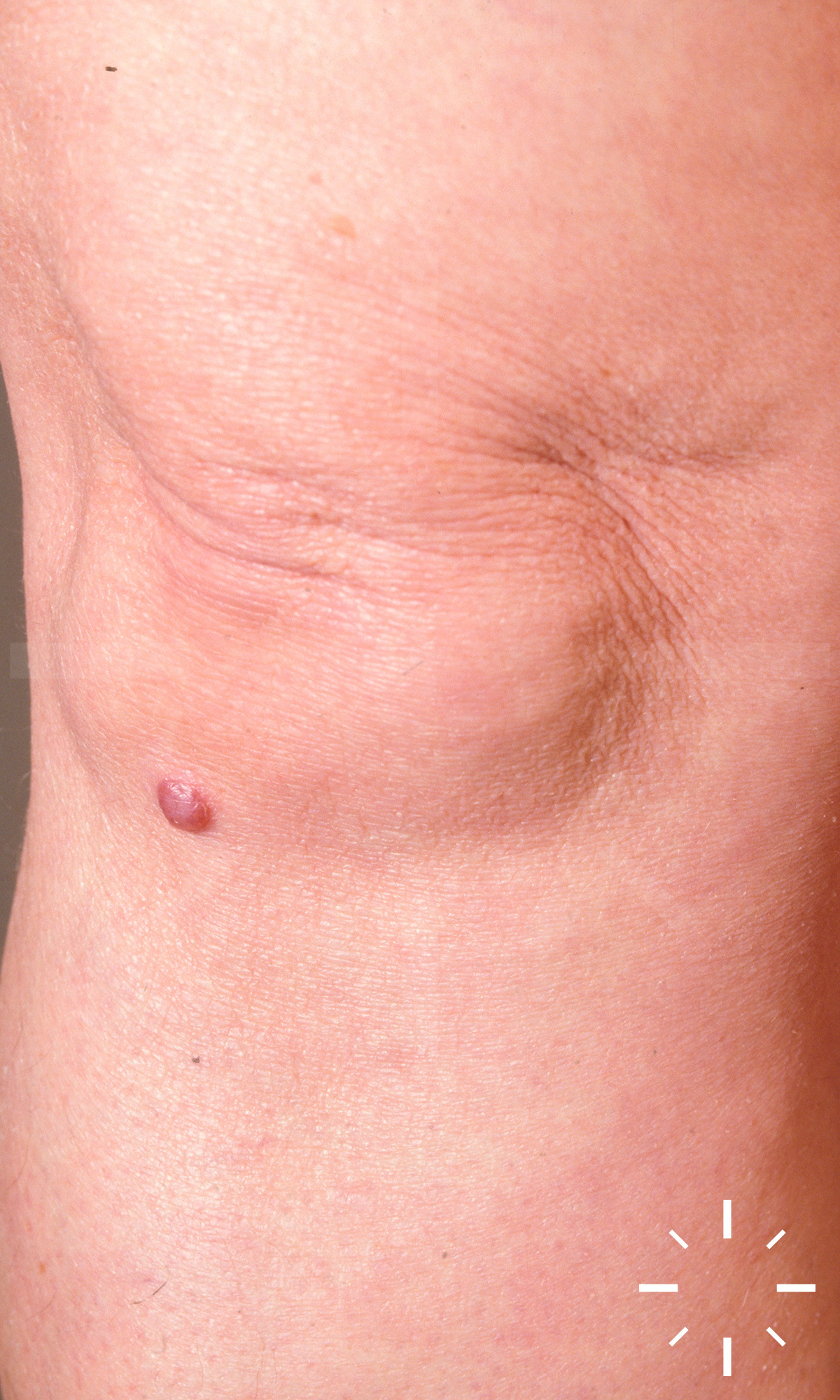
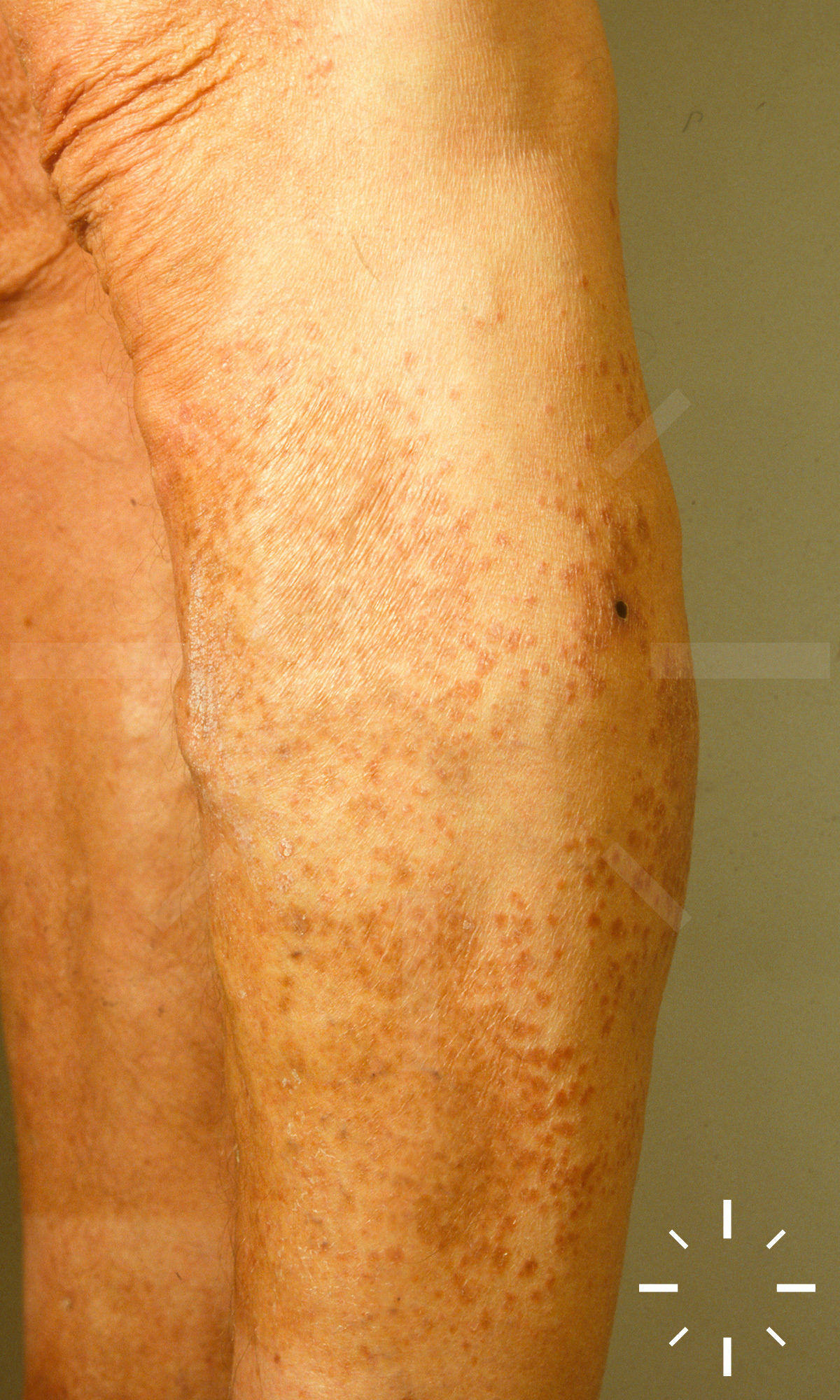
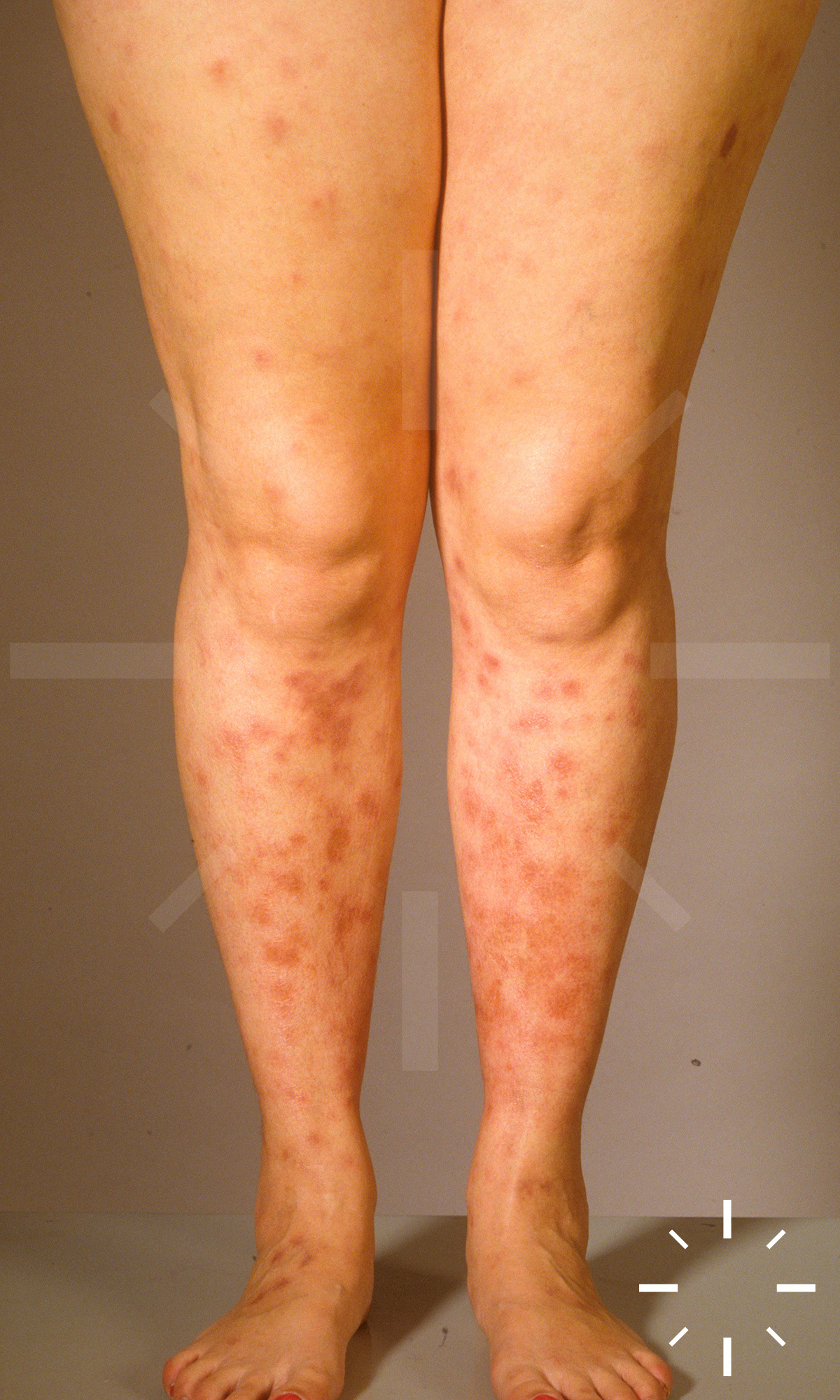
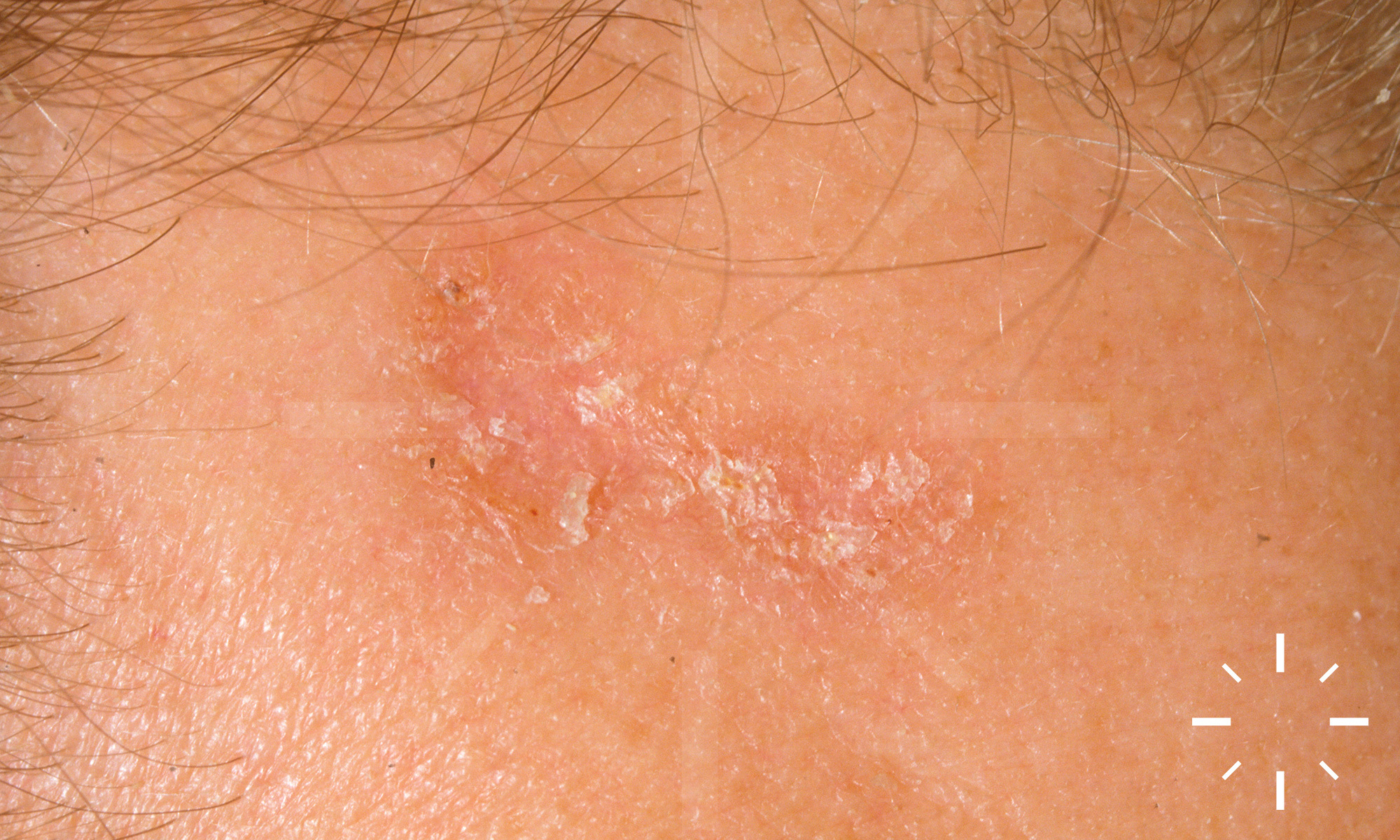
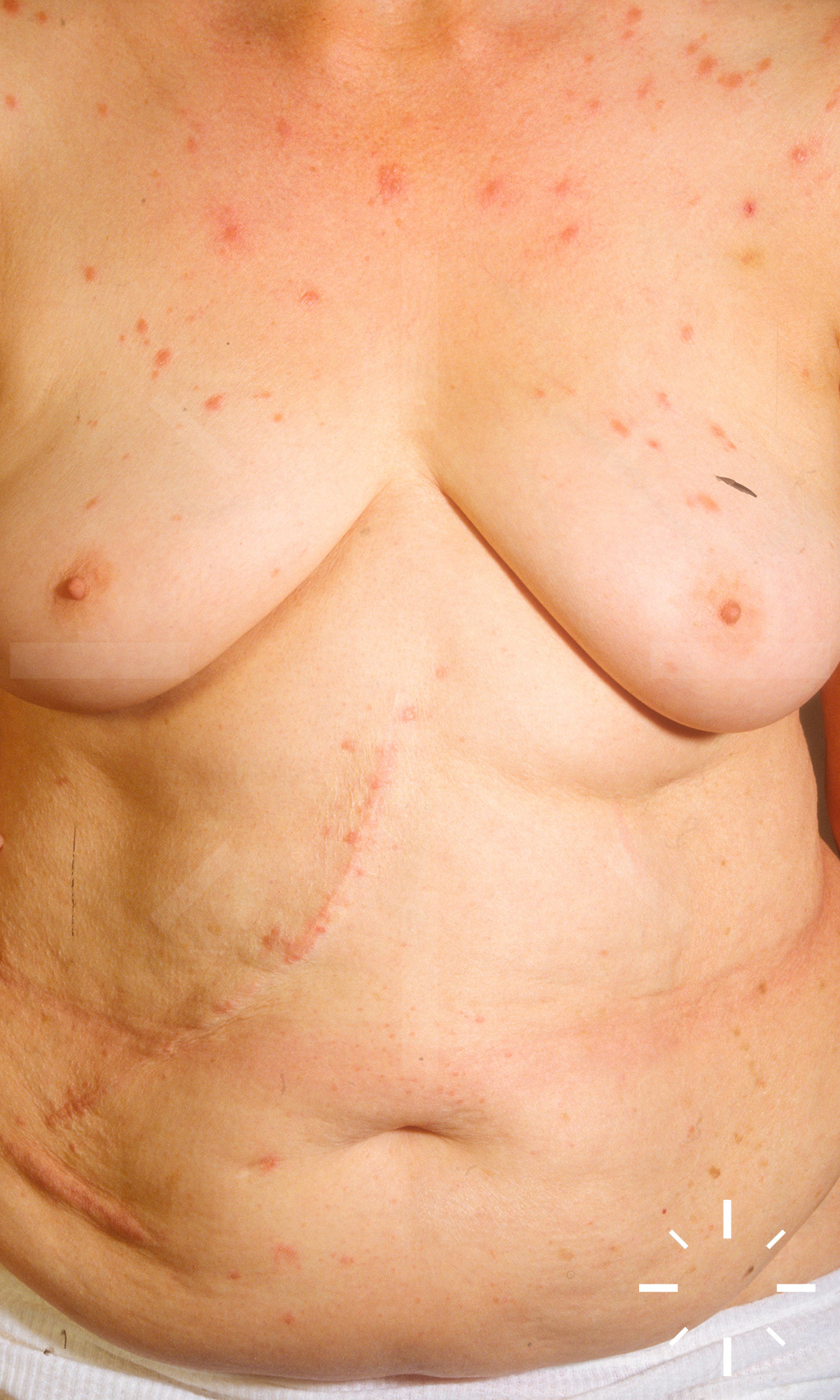
1/60